

Full Length Research Article
Cloning and expression of hepatitis B surface gene in E. coli
Krishma Gulzar1, Maria Sharif1, Arif M Khan1, Muhammad Rizwan Javed2, Imran Riaz Malik1*
Adv. life sci., vol. 8, no. 3, pp. 275-280, July 2021
*- Corresponding Author: Imran Riaz Malik (Email: imran.riaz@uos.edu.pk)
Authors' Affiliations
2. Department of Bioinformatics and Biotechnology (BNB), Government College University Faisalabad (GCUF), Faisalabad – Pakistan
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Hepatitis B virus (HBV) is among the smallest DNA viruses resulting in ~800,000 deaths each year. Pakistan is considered a country affected by HBV. In Pakistan, the most dominant genotype is D. HBV is an enveloped virus of 3.2 kb. The study's goal was to express hepatitis B surface antigen in a bacterial host to produce a recombinant protein.
Method: Blood samples were collected in EDTA coated vacutainer from patients after their consent. DNA was extracted from serum through the phenol-chloroform method; Hepatitis B surface gene was cloned in TA cloning vector, subclone in pET 28a expression vector. An expression vector containing the Surface gene was then transformed into a competent bacterial host BL21 and inducted with IPTG at 0.1-0.2mM concentration for expression. The expressed proteins (soluble and pellet form) were analyzed on SDS PAGE.
Results: Hepatitis B Surface gene of 681bp after PCR were detected under UV light then successfully cloned and subcloned in pET 28 expression vector. The restricted fragment indicating the gene of interest was 681bp when analyzed on 1.2% Agarose gel under UV light. The required protein of 25kDa was obtained in soluble form when detected on 12% SDS PAGE after staining with Coomassie Blue dye.
Conclusion: Hepatitis B surface gene was successfully expressed in both insoluble and pellet forms using E.coli. The expression of surface protein needs to maximize through optimizing conditions to be used as potent candidate for vaccine production to prevent hepatitis B infection.
Keywords: Hepatitis B virus; Surface gene; Cloning vector; pET expression vector
Introduction![]()
Chronic hepatitis B infection is the primary health problem globally. It is fundamentally a leading cause of liver cancer with actual infection load hence interpreting the significant unfulfilled health issues [1]. Generally, blood and blood products carry these viral infections. There is a failure to induce a major innate immune response within liver cells. The incomplete clearance of infected hepatocytes causes immunosuppression in the microenvironment of the liver after the first exposure to the hepatitis B virus [2]. Up to 4% of the world’s inhabitants suffer from chronic hepatitis B . Annually, ~ 800000 people infected with this particular virus are at risk of suffering from liver cirrhosis and hepatocellular carcinoma (HCC) [3].
The HBV can be divided into various genotypes and subgenotypes based on huge genetic variance due to transmission routes, geographical distribution, disease progression, response to vaccination, and clinical outcome measures [4]. Approximately 13 million population of Pakistan were infected by the hepatitis B virus as investigated by the researchers of Pakistan Medical Research Council during 2007- 2008 [5]. In Pakistan, according to the recent analysis, HBV/D is reported to be the most prevalent genotype [6].
The multi-gene eukaryotic expression vector pcDNA3-HBsAg-p3O-ROP2 was established after Hepatitis B surface antigen was amplified and cloned. The constructed expression vector contained complete sequences of the p3O-ROP2 compound gene and HBsAg, and this recombinant expression vector could be used to develop multi-gene nucleic acid vaccines [7]. For the production of vaccines for global use, a prokaryotic expression system, especially E. coli, is considered as the preferred host. Good safety and affectivity of Hecolin, the first licensed virus-like particle vaccine delivered from Escherichia coli, has also been demonstrated [8]. A PCR-amplified DNA encoding Indonesia sHBsAg was cloned into Hansenula polymorpha expression vector pHIPX4. The resulting pHIPX4-sHBsAg was integrated into the alcohol oxidase locus of H. polymorpha NCYC495 genome, sHBsAg was regulated under the control of AOX promoter. Expression of sHBsAg was detected by HBsAg diagnostic kit Tests [9].
E. coli BL21 (DE3) is one of the most accepted systems for the production of genetically engineered protein that is commonly used in combination with pET expression system. Insoluble inclusion bodies were formed by induction of isopropyl-B-D-1-thiogalactopyranoside (IPTG) by stressing the cell [10].
The main objective of this study was the cloning and expression of hepatitis B surface gene using a bacterial host.
Methods![]()
Screening through a diagnostic set of primers
Blood samples of 5cc were collected in EDTA coated vacutainer with the patient's consent from the hepatitis B virus infected individuals admitted in the wards of different hospitals of Sargodha. These patients were examined based on their background history, biochemical parameters, Alanine aminotransferase (ALT), Aspartate aminotransferase (AST) values mentioned on the questionnaire. Viral DNA was extracted from hepatitis B virus using Phenol-Chloroform method as described by (11). Primers were designed for qualitative PCR by using an online tool (Software vector NTI). The following two types of diagnostic set of primers B1: 5-CATCCTGCTGCTATGCCTCATCT-3, B2: 5-CGAACCACTGATCAAATGGCACT-3, B3: 5-GGTATGTTGCCCGTTTGTCCTCT-3 and B4: 5-ACTAGTAAACTGAGCCA-3, were used for the amplification of hepatitis B viral DNA. Two types of Polymerase Chain Reaction were performed, including regular and nested PCR that involves different reagents used.
Regular PCR
A total volume of 50µl of the reaction mixture was prepared in 0.2ml PCR tubes containing 26.5µl double deionized distilled H2O, 3µl of MgCl2, 3µl of deoxynucleotide triphosphate (dNTPs), 0.5µl of Taq DNA polymerase, Taq Buffer 5µl, 1µl of each primer B1, B2 (forward primer and reverse primer), and 10µl of target DNA template.
Nested PCR
A total volume of 50µl of the reaction mixture was prepared in 0.2ml PCR tube containing 34.5µl dd H2O, 5µl of Taq buffer, 5µl of MgCl2, 5µl of deoxynucleotide triphosphate (dNTPs), 0.5µl of Taq DNA polymerase, 1µl each primers B3, B4 (forward primer and reverse primer), and 2µl of target DNA.
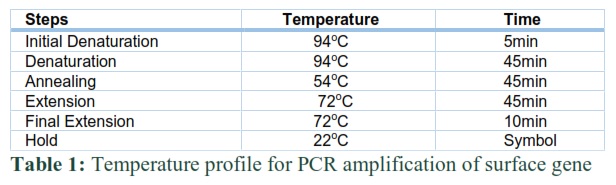
Amplification of surface gene through PCR
Polymerase chain reaction (PCR) was done in 0.2ml PCR tubes contains a total reaction mixture of 50μƖ. The mixture was made by mixing 10μƖ positive DNA template, 5μƖ of Taq Buffer (Fermentas), 3μƖ of MgCl2 (Fermentas), dNTP’s 3μƖ, , Forward primer RHBSF1 and RHBSR1 1μƖ each for regular PCR, Primers HBSF1 and HBSR1 1μƖ each for Nested PCR, 0.5μƖ of Taq polymerase (Fermentas) and 26.5μƖ of PCR water (ddd H2O). The temperature profile used is described (Table 1). The resulted in PCR amplified products of encoding genes were resolved on 1.2% Agarose gel.
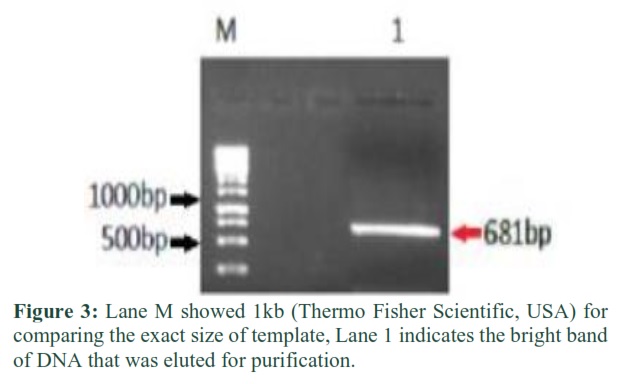
Gel Elution and Purification of DNA
After confirmation through visualization of purified DNA bands through Gel Doc, the agarose gel size containing the purified DNA fragment was excised carefully and placed into a pre weighted tube (mg). The weight of gel slice containing DNA was recorded by excluding the weight of Eppendorf. The DNA is purified through Gene Jet Purification Kit, the purified DNA was confirmed through gel electrophoresis.
Construction of expression vector containing hepatitis B surface gene
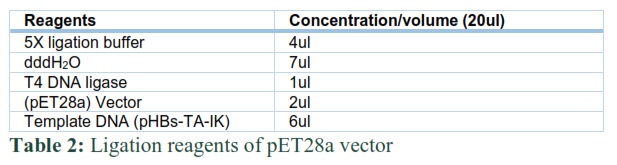
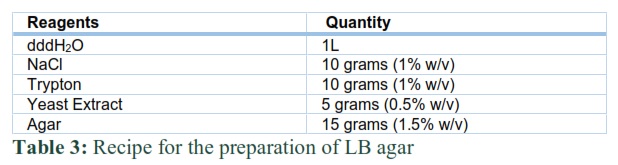
Hepatitis B surface was first cloned in TA cloning vector and then subcloned in expression vector pET28a under the control T7 promoter through restriction digestion (EcoRI and HindIII) method. The ligation mixture is described in Table 2. For the growth of transformed E.coli cells, a nutrient-rich media is required. LB agar appeared as a gel on petri-plates after solidification.
Transformation of plasmid DNA into BL21 cells
The chilled aliquots of the competent cells (BL21) containing the quantity of 100μƖ were frozen at -80oC. For transformation, they were thawed on ice and mixed with 5μƖ of ligation mixture and was placed on ice for 30 min. The cells were heat-shocked at 42oC for 2 min followed by cooling on ice for 2 min, immediately after that 1ml LB broth was added.
Plasmid isolation and confirmation through restriction analysis:
The plasmid was isolated through the kit method (Ferments). The plasmid was confirmed through restriction digestion with EcoRI and Hind III.
Expression analysis (Small scale protein expression) for expression vector containing hepatitis B surface gene
4ml culture containing pET 28a clones was taken and kept in shaking incubator at 220rpm, and temperature maintained was from 28 to 30oC. The next day 4ml fresh culture was taken and inoculated with 0.2ml of overnight culture until OD reached (600nm) 0.6. It was then kept in a shaking incubator for 3 to 4 hours.
IPTG Induction
Each culture had IPTG to 0.1-0.2mM (10ml culture needs 10ul of 100mM stock solution). Shaking was done at 220rpm for 4hours, and pellet was spin down for 10 min at 6000rpm and freeze for storage at -20oC.
Lysis of Bacteria
1ml of lysis buffer was added to the small pellet, and the bacterial pellet was freezed by the addition of lysozymes in a large volume. During lysozyme digestion, enzyme powder was added to the bacterial pellet and kept on ice. This solution was then transferred to a shaking incubator at 30oC for 1 hour. Due to the presence of genomic DNA a highly viscous product was obtained, and the passage reduced this viscosity through 21-gaugeneedle. Later the pellet cell debris was separated by centrifuging it at 6000rpm for 3min and supernatant (protein) was transferred to the new tube. Lysate supernatant was precipitated with 80% ammonia sulfate for overnight at 4oC. The precipitated protein was spun down for 30 min at 14000 rpm and about 50-100mgs of protein was obtained per liter.
Analysis of the expressed protein through SDS-PAGE
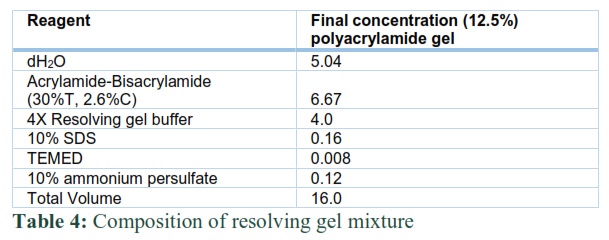
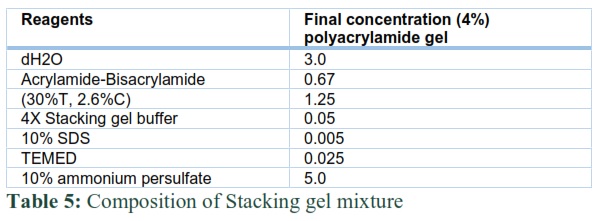
The mass of subunits was determined by applying the expressed protein on 12% SDS PAGE. The resolving gel mixture (Table 4) was transferred into the gel apparatus which was separated by sandwiching 2 spacers between two glass plates. After pouring it, n-butanol was layered on the top of the gel to get good surface. Then n-butanol was removed from the top of the gel by washing it many times with dH2O. Later the stacking gel mixture (Table 5) was poured on the top of polymerized resolving gel. Combs were inserted and the gel was allowed to cool and polymerize. Before the loading of expressed protein into the gel, it was mixed in 2X samples of loading buffer for 3 to 5 min for binding of SDS to the enzymes. A protein marker with
The molecular marker range in size from 10kDa to 200kDa was also run along the gel to measure the size of desired recombinant protein.
SDS PAGE was run first on 80 volts and then on 120 volts. As the tracking dye reached at the middle or bottom of the gel, it was dipped in fixing solution for 10 min at room temperature having 50% methanol and 10% v/v glacial acetic acid on dH2O, then the gel was stained with Coomassie brilliant blue R-250 stain (50% v/v methanol, 10% v/v glacial acetic acid and 0.1% v/v Coomassie brilliant blue R-250 in a destaining solution) (40% v/v methanol and 10% v/v glacial acetic acid in distilled water) until the background was clear.
Results![]()
More than 80 blood samples were collected from hepatitis B infected patients and serum was extracted in the present study. The DNA was extracted with the help of Phenol-Chloroform extraction method. The surface gene was amplified through PCR and then cloning was done in T/A vector. It was later sub-cloned in pET28 and expression was analyzed through SDS PAGE.
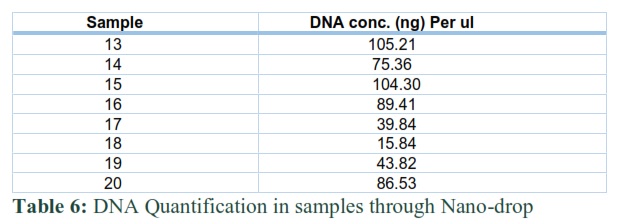
DNA Quantification
DNA was extracted from the serum samples infected with HBV using phenol chloroform extraction method. The HBV DNA concentration was also determined using nano-drop prior to amplification through PCR. The concentrations of samples are listed in a table below:
Screening of Hepatitis B virus patients through PCR
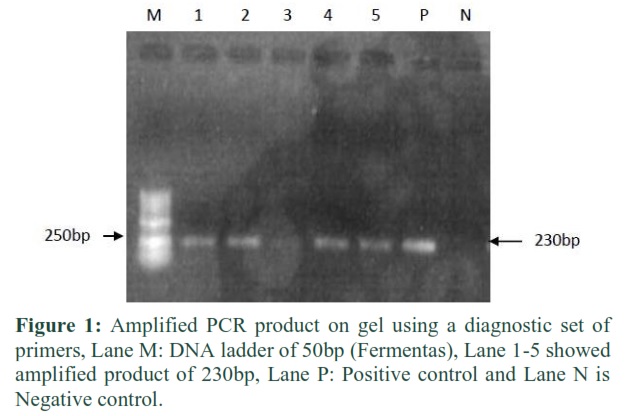
Hepatitis B virus patients screening was done by using diagnostic set of primers and the amplified DNA was detected on 1% agarose gel as shown in Figure 1 with band size of 230bp compared with 50bp DNA marker.
Hepatitis B surface gene amplification through PCR
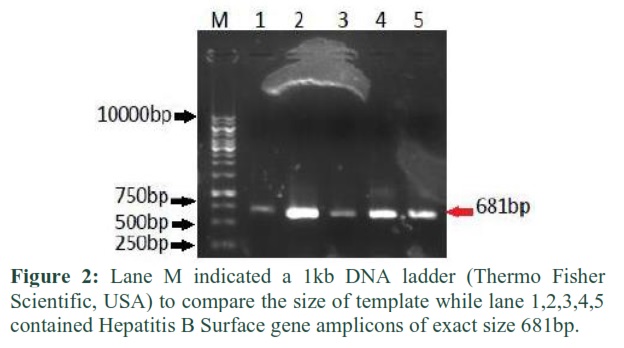
Through PCR by using specific reconstituted primers and control (positive control and negative control) hepatitis B surface gene was amplified, and these PCR amplicons were detected under UV light in gel doc of Syngene. The best results of hepatitis B surface gene amplification examined on 1.2% agarose gel are shown in Figure 2.
Alhusseini et al., reported the same the results of HBV DNA amplification indicating the same band size of 681bp as indicated by present study [12]. A local recombinant sHBsAg was produced, a PCR amplified DNA fragment encoding Indonesia sHBsAg was cloned in to Hansenula polymorpha expression vector pHIPX4 [9].
Gel Elution
The PCR product was loaded on gel and analyzed under UV light of gel doc after confirmation of template using cutter the gel slice was excised and eluted from the gel and purified (Figure 3). Bandehpour et al., showed the same results of gel elution after the confirmation using agarose gel and HBV DNA with 681bp visualized which was needed for the purpose DNA purification [13].
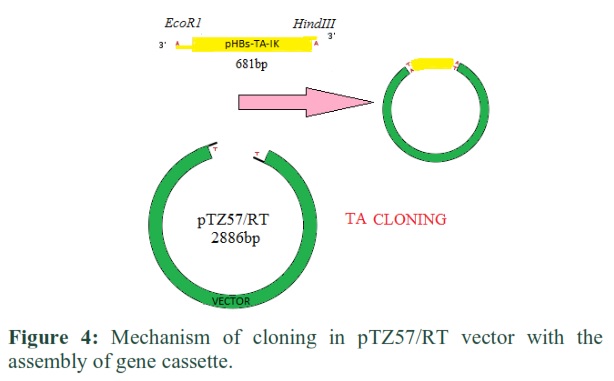
Cloning strategy for the creation of expression cassette under T7 promoter in pTZ57/RT Vector containing hepatitis B surface gene
A T/A cloning vector and gene of interest were restricted by using two endonuclease enzymes (EcoR1 and Hind III) later the surface gene of 681bp was ligated in pTZ57/RT vector having the size of 2886bp. The size of the recombinant gene (pHBs-TA-IK) obtained was equals to 3567bp (Figure 4).
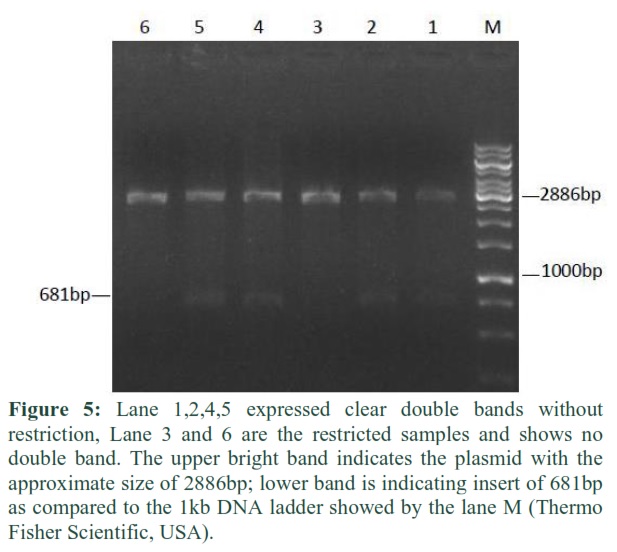
Restriction Analysis
The pTZ57/RT cloning vector was double digested using restriction enzymes EcoR1 and Hind III and their specific buffers. The cloning was confirmed in T/A vector and observed under gel documentation system after running it on agarose gel through restriction analysis. Exclusively Hepatitis B surface gene clone was confirmed by using restriction enzymes and compared against 1kb general to estimate the required sized band (Figure 5).
In a study, the recombinant plasmid pHIPX4-sHBsAg was verified by restriction enzyme analysis using Hind III and Sal1.The digested plasmid gave two fragments which represented the Phipx4 vector and sHBsAg insert [9]. After digestion the plasmid was isolated and purified in order to ligate it with the same enzymes to obtain the expression vector [14].
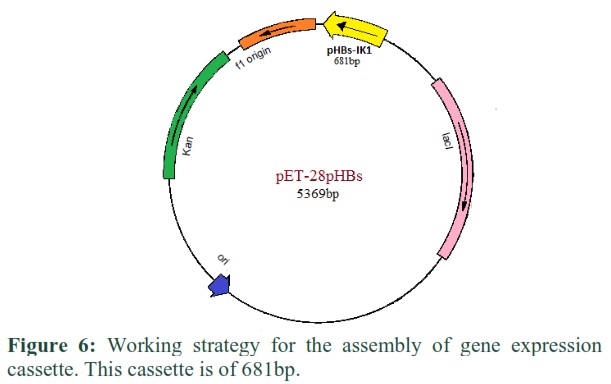
Cloning strategy for the creation of expression cassette in pET-28pHBs Vector containing hepatitis B surface gene
The Pet28 vector and surface gene (TA clone) were restricted with the same enzymes (EcoR1 and HindIII) and were ligated together after restriction analysis. The recombinant gene (pHBs-IK1) obtained was the size of 6050bp containing the size of gene of interest equals to 681bp and the size of expression vector was 5369bp (Figure 6).
Protein Expression analysis (SDS-PAGE)
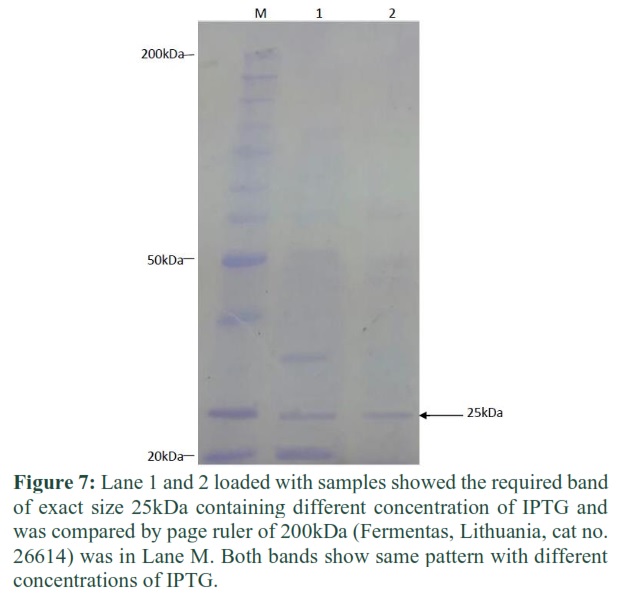
By applying the purified proteases on 12% SDS PAGE the subunit molecular mass was determined. SDS-PAGE was run at a voltage of 80 volts and then of 120 volts. The PAGE was stopped when tracking dye front reached at the bottom of the gel. The required expressed protein band size observed was of protein was 25kDa (Figure 7).
The plasmid mu-IFN-CSP/pET-21b was transformed into E.coli BL21 (DE3) for protein expression. After induced with IPTG (1.0 mM/L culture) for 4 hours, the recombinant protein with approximately similar size was successfully expressed with 75.2% solubility [15].
Prethrombin-2 was expressed in E.coli ER2566 by induction of various IPTG concentrations at 12oC. 63kDa protein band obtained from the soluble fraction on SDS-PAGE indicates that IPTG concentration at 0.010mM was the suitable IPTG concentration [16].
Figures & Tables
Discussion![]()
Chronic hepatitis B infection is the main health problem globally and is fundamentally a main cause of liver cancer with actual infection load hence interpreting the large un-fulfilled health issues. E. coli BL21 (DE3) is one of the most accepted systems for the production of genetically engineered protein that is commonly used in combination with pET expression system. Insoluble inclusion bodies were formed by induction of isopropyl-B-D-1-thiogalactopyranoside (IPTG) by stressing the cell [10].
The main purpose of this study was cloning and expression of hepatitis B surface gene in E.coli. The DNA was extracted with the help of Phenol-Chloroform Extraction method. After successful PCR amplification of surface gene and cloning in T/A vector. It was sub-cloned in pET28 and expressed protein was analyzed through SDS PAGE.
Hepatitis B virus patients screening was done by using diagnostic set of primers and the amplified DNA was detected on 1% agarose gel with band size of 230bp.
The amplified hepatitis B surface gene of 681bp were detected on 1.2% agarose gel which are in agreement as Alhusseini et al., reported the same the results of HBV DNA amplification indicating the same band size of 681bp [12]. A local recombinant sHBsAg was produced,a PCR amplified DNA fragment encoding Indonesia sHBsAg was cloned in to Hanusenula polymorpha expression vector pHIPX4 [9].
A T/A cloning vector and gene of interest were restricted by using two endonuclease enzymes (EcoR1 and Hind III) later the surface gene of 681bp was ligated in pTZ57/RT vector having the size of 2886bp. Exclusively Hepatitis B surface gene clone was confirmed by using restriction enzymes and compared against 1kb generuler to estimate the required sized band. The result are in agreement with study carried out by Heryakusuma et al., [9]. The recombinant plasmid pHIPX4-sHBsAg was verified by restriction enzyme analysis using Hind III and Sal1.The digested plasmid gave two fragments which represented the Phipx4 vector and sHBsAg insert [9]. After digestion the plasmid was isolated and purified in order to ligate it with the same enzymes to obtain the expression vector [14].
The pET28a vector and surface gene (TA clone) were restricted with the same enzymes (EcoR1 and HindIII) and were ligated together after restriction analysis. The recombinant gene (pHBs-IK1) obtained was the size of 6050bp containing the size of gene of interest equals to 681bp and the size of expression vector was 5369bp.
By applying the purified proteases on 12% SDS PAGE the subunit molecular mass was determined. SDS-PAGE was run at a voltage of 80 volts and then of 120 volts. The required expressed protein band size observed of protein was 25kDa. The plasmid mu-IFN-CSP/pET-21b was transformed into E.coli BL21 (DE3) for protein expression. After induced with IPTG (1.0 mM/L culture) for 4 hours, the recombinant protein successfully expressed with 75.2% solubility [15]. Prethrombin-2 was expressed in E.coli ER2566 by induction of various IPTG concentrations at 12oC. The results showed that IPTG concentration at 0.010mM was the suitable IPTG concentration [16].
Author Contributions
KG and MS carried out research work, IRM design research proposal, supervised research work and Manuscript preparation, RJ supervised research work and draft preparation, AMK help in draft preparation and samples collection.
It is declared by the authors that they have no conflict of interest about the research paper or any of its part.
Acknowledgement
This research work was funded by ORIC, University of Sargodha.
References![]()
- Tang CM, Yau TO, Yu J. Management of chronic hepatitis B infection: current treatment guidelines, challenges, and new developments. World Journal Gastroenterology, (2014); 20: 6262-6278.
- Guidotti LG, Isogawa M, Chisari FV. Host-virus interactions in hepatitis B virus infections. Immunology, (2015); 36: 61-66.
- Komatsu H. Hepatitis B virus: where do we stand and what is the next step for eradication? World Journal Gastroenterology, (2014); 20: 8998-9016.
- Pourkarim MR, Olyaee SAB, Kurbanov F, Ranst MV, Tacke F. Molecular identification of hepatitis B virus genotypes/subgenotypes: Revised classification hurdles and updated resolutions. World Journal of Gastroenterology, (2014); 20 (23): 7152-7168.
- Farhat M, Yasmeen A, Ahmad A. An overview of Hepatitis B and C in Pakistan. Journal of Microbiological Allied Sciences, (2014); 1(2): 98-102.
- Majid M, Razza A, Anwar MA, Qayyum M, Zaman N, Khanum A, Beg MA. Analysis of Complete and Partial Genome Sequences of Hepatitis B Virus and Determination of its Genotypes and Sub-Genotypes from Pakistan. Pakistan Journal of Zoology, (2016); 48(3): 747-753.
- Wei QK, Xiao T, Li J, Yin K, Jia F, Xu C, Zhao G, Cui Y, Liu G, Sun H, Jiang H, Yan G, Huang B. Construction and identification of Complex DNA vaccine of hepatitis B and Toxoplasma gondii. International Journal of Clinical and Experimental Medicine, (2015); 8(6): 9156-9161.
- Huang CH, Chen E, Yang YY, Chen YY, Chang JJ, Chen KJ, Lu CH, Lee KD, Chen PC, Chen CC. The impact of hepatitis B virus infection and vaccination on the development of non-Hodgkin lymphoma. Journal of Viral Hepatitis, (2017); 10(24): 885-894.
- Heryakusuma C, Puspasari F, Ihsanawati, Arifin E,Rachman G, Irasoniatan M, Ramadhani E, Nurainy N, Natalia D. Cloning and Expression of small Hepatitis B Surface Antigen (sHBsAg) in Hansenulla polymorpha. Microbiology Indonesia, (2016); 10: 119-124.
- Wurm DJ, Veiter L, Ulonska S, Eggenreich B, Herwig C, Spadiut O. The E.coli pET expression system revisited mechanistic correlation between glucose and lactose uptake. Applied Microbiology and Biotechnology, (2016); 100(20): 8721-8729.
- Malik A, Kumar D, Khan AA, Khan AA, Chaudhary AA, Husain SA, Kar P. Hepatitis B virus precore G1896A mutation in chronic liver disease patients with HBeAg negative serology from North India. Saudi journal of biological sciences, (2018); 25(7):1257-62.
- Alhusseini NF, Abadeer MZ, El-Taher SM. Hepatitis B virus DNA can be amplified directly from blood spot on filter paper. American Journal of Biochemistry and Biotechnology, (2012); 2(8): 143-149.
- Bandehpour M, Khodabandeh M, Kazemi B. Cloning and Expression of Hepatitis Surface Antigen. Hepatitis Monthly, (2008); 8(1): 17-21.
- Zhang R, Xu Y, Xiao R, Wang S, Zhang B. Improved production of (R)-1-phenyl-1,2-ethanediol using Candida parapsilosis (R)-carbonyl reductase expressed in Pichia pastoris. Process Biochem. (2011); 46(3): 709-713.
- Lui A, Gui S, Zhang L, Chen Z, Tang Y, Xiao M, Wang J, Liu W, Jin X, Zhu J, Lu X. Priduction of Bioactive-Liver targeting interferon Mu-IFN-CSP by soluble prokaryotic expression. AMB Express, (2017); 7: 192
- Rizkia PR, Silaban S, Hasan K, Kamara DS, Subroto T, Soemitro S, Maksum IP. Effect of Isopropyl-β-D-thiogalactopyranoside concentration on prethrombin-2 recombinan gene expression in Escherichia coli ER2566. Procedia Chemistry, (2015); 17: 118-24.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0


