

Full Length Research Article
Density function theory (DFT), spectroscopic, and molecular docking studies of an antidepressant drug
Naved Azum*, Muhammad Nadeem Arshad, Malik Abdul Rub, Mohammad Asad, Khalid Ahmed Alzahrani, Hadi M Marwani
Adv. life sci., vol. 12, no. 1, pp. 175-184, February 2025
*- Corresponding Author: Naved Azum (Email: navedazum@gmail.com, nhassan2@kau.edu.sa)
Authors' Affiliations
[Date Received: 02/09/2024; Date Revised: 05/12/2024; Date Published: 31/12/2024]
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Amitriptyline is specifically recommended for the treatment of anxiety that is associated with depression. Since 1950, authorities have authorized its use for the treatment of severe mood disorders. One of amitriptyline's main antidepressant properties is its ability to block the central nervous system's serotonin and norepinephrine absorption.
Methods: In this study, we computed the quantum computation data for amitriptyline (AMT) using density function theory (DFT) and molecular docking techniques. We evaluated the shifts in H-NMR and 13C-NMR using the GIAO approach and compared the obtained results with the experimental spectra. We conducted molecular docking, using two receptors to identify the most optimal interactions between ligands and proteins.
Results: The study showed that the geometrical parameters (bond lengths, angles, and torsion angles) from DFT were very close to the experimental data, with an average discrepancy of 0.01 Å to 0.03 Å. We obtained comparable experimental and theoretical spectroscopic data. We performed molecular docking investigations on the 4IB4 and 8IRV proteins.
Conclusion: This study investigates the properties of AMT at the quantum mechanical level. This paper meticulously investigates several significant parameters, comparing calculated spectroscopic results (UV, FTIR, HNMR, and CNMR) to experimental data. This study demonstrates the specified molecule's non-linear optical properties. When docking the molecule with the two proteins, we calculate the binding energy to be -8.9 and -9.6 kcal/mol, indicating that the compound merits further investigation for its medicinal applications.
Keywords: Amitriptyline; Density function theory; Molecular docking; MEP; ELF
Introduction![]()
In the field of psychiatry, tricyclic antidepressants have been extensively utilized for the treatment of a variety of disorders, with a particular emphasis on clinical depression [1–3]. In most cases, the primary objective of these medications is to inhibit the uptake of norepinephrine or serotonin in the presynaptic region. However, the potency of these medications varies, and they tend to cause unwanted side effects. Although there are more recent and less dangerous alternatives, tricyclic antidepressants continue to be prescribed because of their lower cost and reputation as the most prominent class of antidepressants. This is the case even though there are other options available. Three rings of atoms make up the molecular structure of tricyclic antidepressants, where the name of these medications comes from [4–7]. In most cases, the core ring is made up of seven atoms, and the side chain is made up of either N-alkyl methylamine or N-alkyl dimethylamine. Drugs such as imipramine, desipramine, clomipramine, amitriptyline, nortriptyline, doxepin, and trimipramine are examples of tricyclic antidepressants that are commonly used [8–10].
As a tricyclic antidepressant, amitriptyline is especially prescribed for the treatment of depression, both endogenous and psychotic, as well as the anxiety that is inherently connected with depression. For the treatment of serious affective disorders, it has been approved for use since the year 1950 [11]. The capacity of amitriptyline to inhibit the uptake of serotonin and norepinephrine in the central nervous system is primarily responsible for its antidepressant effects. On the other hand, it also possesses additional pharmacological properties, such as the ability to block histamine, muscarinic, alpha-1 adrenergic, and serotonin receptors, in addition to various ion channels, even when present in extremely low concentrations [12,13]. Several in vivo models linked to acute, local delivery of low doses of amitriptyline have demonstrated peripheral anti-inflammatory and analgesic activities in addition to its antidepressant effects. There is a probable chance that its pharmacological activities are responsible for the analgesic and anti-inflammatory effects that it possesses. Amitriptyline has little propensity for inhibiting the uptake of adenosine, a purine-based medication that is effective in treating pain and inflammation.
AMT has a ring structure, and the groups (10,11-dihydro-5H-dibenzo[a,d] annulene and 3-(dimethylamino propylidene) are the components that make up the molecule. A total of 46 bonds are present in the molecule of AMT. A total of twenty-three non-H bonds, thirteen multiple bonds, three rotatable bonds, one double bond, twelve aromatic bonds, two six-membered rings, one seven-membered ring, two eleven-membered rings, and one tertiary amine (aliphatic) are present in the sample.
A growing number of researchers in the various fields of science are beginning to recognize the significance of the area of computational chemistry [14]. Estimating the geometrical characteristics of compounds is an important part of organic chemistry since it helps to understand the molecular structure and provides insights into reaction pathways and mechanisms [15–17]. In addition, it plays a vital role in the field of organics. It is also possible for computational studies to provide extensive information regarding the electrical characteristics of reactants, intermediates, and products, enabling research to be compared with various experimental studies [18,19]. The growing collaboration between experimental and computational chemistry has played a crucial role in solving numerous problems organic chemists face. Additionally, DFT (Density Function Theory) has recently achieved significant synthesis chemistry advancements [20,21]. Both chemistry and physics extensively utilize the quantum-mechanical (QM) technique known as density functional theory. This tool (DFT) allows for calculating the electronic structures of atoms, molecules, and solids. The field of computational solid-state has widely used this technique since 1970. It was not, however, widely used in quantum-chemical applications until the 1990s, after considerable modifications were made to the approach, which resulted in improved accuracy. DFT is known for having a better price-to-performance ratio when compared to other wave function-based methods, like Møller-Plesset perturbation theory or coupled cluster [22]. Because of this, it is possible to research molecular systems that are more significant and useful with appropriate accuracy. This has extended the predictive potential inherent in electronic structure theory. As a result, DFT has become the most commonly used electrical structure method. In addition, computational chemistry has the potential to generate fresh concepts for the development of novel chemical reactions and the execution of mechanistic research.
Since AMT has a significant therapeutic use, we decided to do theoretical research on AMT compounds (DFT/TD-DFT and Molecular Docking). We utilized the DFT and TD-DFT methods, along with the B3LYP functional and 6-311++ G (d, p) basis sets, to determine the most appropriate geometry parameters for the molecule. Additionally, we looked at the possibility of correlations between the data at hand and the theoretical framework.
Methods![]()
Spectroscopic studies
We recorded the NMR spectra using a Bruker ultra shield plus 600 spectrometers operating at a proton resonance frequency of 600 MHz. We created the solutions for AMT using deuterium oxide (D2O). We transferred approximately 1 millilitre of the solution to a 5-millimeter NMR tube for NMR spectra. We recorded the chemical shifts using the ppm scale (Table 1). We used the compound tetramethyl silane (TMS) as an internal standard. The chemical shift reading had a precision of approximately ±0.01 ppm. The calculated relative uncertainties for chemical shifts (δ, ppm) are 4%. The UV spectra of the AMT were acquired by electronic absorption tests (Figure 1). This experiment employed the Evolution 300 UV-visible spectrophotometer. The UV spectra of the solution were measured in the wavelength range of 200 to 400 nm. The NICOLET iS50 FT-IR spectrometer from Thermo Scientific (Madison, USA) was utilized to conduct FTIR studies (Figure 1). A spectrum of pure H2O was subtracted from the system under investigation. Only a portion of the wavelength is displayed to maintain clarity in the graph.
Computational analysis details
The density functional theory (DFT) was utilized to achieve complete optimization of the molecular structure of the AMT in its ground state by using Gaussian 09 software [23]. For this purpose, the B3LYP function was used in conjunction with the 6-311++G(d,p) basis set. The molecular electrostatic potential map (MEP) was drawn using the iso-surface of the electron density, which was found to be 0.004 electrons per Å3 (Figure 2). The SCRF-TD-DFT/B3LYP/6-31++G(d,p) was utilized to estimate the electronic absorption spectrum. To figure out how reactive the compound was, we used the frontier molecular orbital (FMO) connected to the highest molecular orbital (HOMO) and the lowest molecular orbital (LUMO). We utilized the Gaussian computer package and the Gauss-View molecular visualization application to perform all the above computations. To conduct topological analysis, an illustration of the electron localization function (ELF) was created using the atoms in molecules (AIM) theory (Figure 2), with the assistance of the Multiwfn program software[24]. Furthermore, molecular docking analyses were effectively performed using Autodock-Vina and Chimaera software, revealing the precise interactions between the molecules. The Chimera program, created at the University of California, San Francisco, enables interactive visualization and study of molecular structures and associated data [25]. This includes supramolecular assemblies, density maps, docking results, sequence alignments, trajectories, and conformational ensembles.
Results![]()
Optimization Investigation
For this particular task, the density function theory (DFT) with B3LYP function, 6-311++ G (d, p) basis set, was utilized [26,27]. As discussed, the AMT structure was optimized in the gas phase using B3LYP density functionals. A frequency calculation followed this to check that the molecule had a conformation with the lowest possible energy. This allowed us to acquire the theoretical IR spectra, which were then compared with the experimental data.
MEP, Molecular electrostatic potential investigation
The molecular electrical potential (MEP) surface can visually represent the charge distributions of molecules in 3D. We can employ the presence of charged regions in a molecule to determine molecular interactions and chemical bond properties. A colour grading system can achieve the visual representation of molecular size, shape, and electrical potential (negative, positive, and neutral), which is of utmost importance. Therefore, by utilizing this knowledge, one can examine the physicochemical characteristics of a molecule [28–30]. Following is the equation that was used to determine the value of V (r):
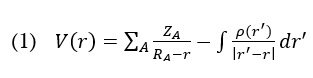
The colours correspond to different levels of electrostatic potential on the AMT's surface (Figure 2). The electrostatic potential grows progressively, from red to orange, yellow to green to blue. The map was colour-coded with a range of -0.039 au (deepest red) to 0.039 au (deepest blue). Red represents the highest level of repulsion, specifically electrophilic assault, whereas blue represents the highest level of attraction, specifically nucleophilic attack [31].
Frontier molecular orbital (FMO) analysis
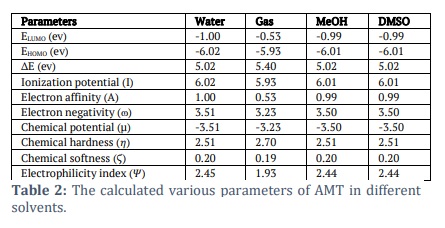
We used the B3LYP/6-311++G (d, p) basis sets to determine the AMT electric and optical properties based on the FMO theory. There exist two distinct categories of molecular orbitals: LUMO (lowest molecular orbitals) and HOMO (highest molecular orbitals [31]. The HOMO orbital, which is the outermost electron orbital, has the potential to function as an electron donor. The energy level of the LUMO corresponds to that of the innermost unoccupied orbital. The LUMO functions as a receptor for accepting electrons [32,33]. The electron-donating capacity, the highest occupied molecular orbital energy (EHOMO), directly influences the molecule's capability to donate electrons. A molecule with a higher HOMO energy (a more negative value) can donate a more significant number of electrons. The energy difference (ΔE) between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is shown in Table 2. The softness, electronegativity, chemical hardness, and electrophilicity index of AMT [34] are in Table 2. The disparity in energy levels between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is indicative of the molecule’s chemical strength and reactivity [35]. The symbol ΔE denotes the energy difference between the HOMO orbital and the LUMO orbital. Table 2 shows the values for the softness, electronegativity, chemical hardness, and electrophilicity index of AMT. This observation makes it clear that these moieties can transition electrons. The energy gap (ΔE) that exists between the HOMO and LUMO orbitals is responsible for the chemical strength and reactivity of the molecule. Our system has a difference in energy gap (ΔE) of 5.399 eV for the AMT. Conversely, compound reactive nature and stability are increased because of the energies within the narrower band space gap. The equations from 2 to 6 showed how the compounds were described in terms of how they reacted, how hard they were chemically, how soft they were, how electronegativity worked, and how electrophilicity worked [36–38].
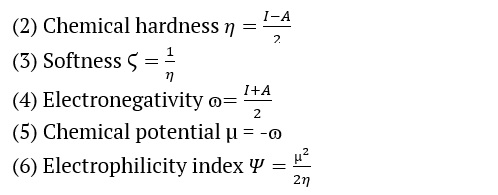
Where A and I are the electron affinity and ionization potential
A = –ELUMO and I = –EHOMO
Population analysis & Fukui Function
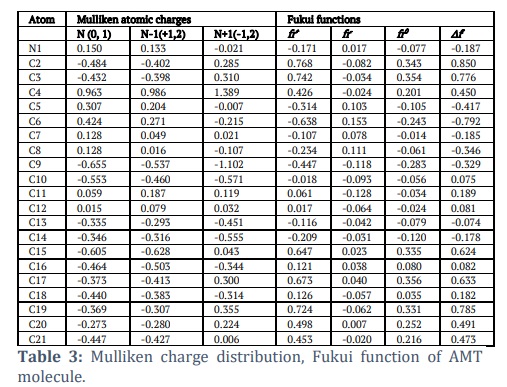
The Mulliken population analysis is a highly effective method for logically explaining variations in electronegativity among atoms inside a molecule. It is frequently employed to substantiate the mapping of molecular electrostatic potential contours [39]. The MEP (Molecular Electrostatic Potential) and Mulliken population were employed to forecast the behaviour of various chemical systems in electrophilic and nucleophilic reactions. Determining effective atomic charges, which represent the charges assigned to each atom and the distribution of positive and negative charges inside molecules, is highly important for manipulating the bond length between atoms. Atomic charges play a crucial role in various molecular phenomena, including the creation of dipole moments, molecule polarizability, electronic structure, acidic and basic behaviour, molecular reactivity, electrostatic potential surface, and other features of molecular systems[40-42]. The Mulliken population inquiry used the B3LYP function and the 6-311++G(d,p) basis set. The findings are presented in Table 3. The nitrogen atom (N1) was regarded as a fundamental site and exhibited a positive atomic charge. The charge distribution indicated that the hydrogen atoms were predominantly concentrated in positive charge regions. Based on the information provided by Table 3, it can be inferred that N1, C4, C5, C6, C7, C8, C11, and C12 possess a positive charge, while C2, C3, C9, C10 and C13 to C21 possess a negative charge. Electronegative atoms are atoms with a negative charge. Introduce an electron-donating group to the carbon atom at position C15, resulting in a negative charge. The charge on the hydrogen atoms of the molecule under study exhibited minor fluctuations because of their attachment to neighbouring atoms. The hydrogen atoms within the examined molecule play a significant role in establishing a hydrogen-bonding network in a crystalline state.
The AMT compound underwent Fukui function study, as the Fukui function is a crucial metric for determining the compound's nucleophilic and electrophilic activity. The Fukui functions' charge values can be found using MPA (Mulliken population analysis). The calculation of Fukui functions can be performed using the following formulas:
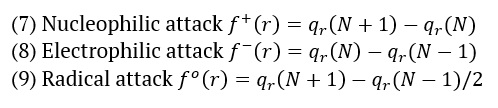
Where qr is the charge of the atom at the rth atomic site, (N) is neutral, (N+1) is anionic, (N-1) is cationic chemical species. The equation can calculate the dual descriptor
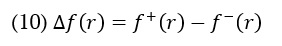
If the value of is positive, it indicates an assault by a nucleophile, whereas a negative value predicts an electrophilic attack. Table 3 presents the Fukui function values for "electrophilic attack , "nucleophilic attack , and radical attack .
Electron localization function (ELF)
This study aims to evaluate the importance of ELF (Electron Localization Function) using Pauli repulsion. When the ELF value is 1, we refer to the related region as the greatest Pauli repulsion region, visually represented by red. Alternatively, when the ELF value is zero or nearly zero, we refer to the area as the minimal Pauling repulsion region, visually represented by the colour blue. The maximum Pauling repulsion corresponds to well-localized electrons, whereas the minimum Pauling repulsion corresponds to delocalized electrons. We can study these concepts in relation to atomic shells, chemical bonds, and lone pairs. ELF analysis employs a quantitative examination of aromaticity to provide essential information about chemical structure, molecular bonding, and reactivity [43,44]. Figure 2 depicts the three-dimensional graph of the compound's electron localization function (ELF). The colour red symbolizes the maximum value of ELF, while yellow to green indicates the intermediate range of ELF values. On the other hand, the colour blue represents the minimum quantity of ELF.
Spectroscopic studies
HNMR and 13CNMR studies
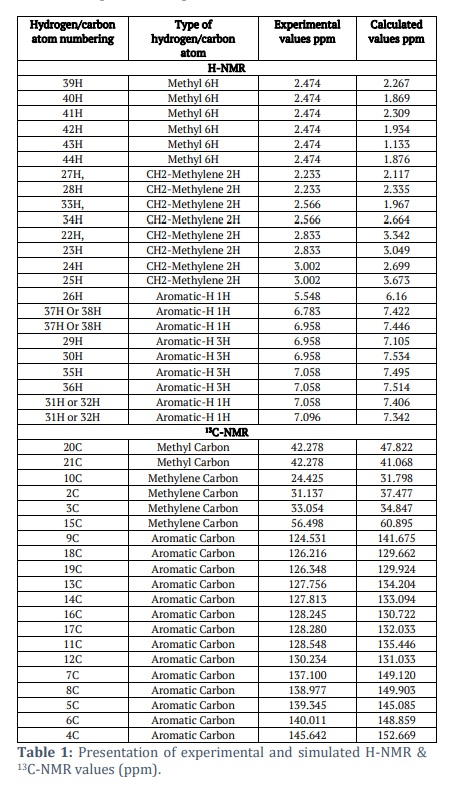
The proton and carbon NMRs of the suggested drug molecule has been studied experimentally [45,46] and computationally. Both the data sets have been tabulated in the Table 1. We have observed a well-established argument between the experimental and simulated values. The structure of the entitled compound was well characterized by the NMR studies.
There are nine aromatic protons found in the region of 5.5 – 7.2 ppm. The aromatic protons appeared at 5.548 ppm, which is expected at H26. The peak at 6.783 ppm is representing the availability of a proton. The two multiplets at 6.958 and 7.058 ppm indicating the presence of 6 protons with the occupancy of 3 protons at each position. The ninth aromatic proton appeared at 7.096 ppm as a triplet. The aromatic protons (26H, 29H, 30H, 31H, 32H, 35H, 36H, 37H, 38H) observed in the simulated data at 6.16, 7.105, 7.342, 7.406, 7.422, 7.446, 7.495, 7.514, 7.534 ppm. The range observed in the simulation data for the aliphatic methyl and methylene hydrogen atoms is 1.133-3.673 ppm while in experimental data these hydrogen atoms appeared in the range of 2.233-3.002 ppm. A singlet observed at 2.474 ppm indicating the 6 hydrogen atoms for two methyl groups in experimental spectrum while these six hydrogen atoms are being expected at the positions 1.133, 1.869, 1.876, 1.934, 2.267 and 2.309 ppm respectively. The methylene protons are identified at 2.233, 2.566, 2.833 and 3.002 ppm with the occupancy of two protons at each position in experimental data while these eight protons appeared at 1.967, 2.117, 2.335, 2.664, 2.699, 3.049, 3.342 and 3.673 ppm.
The compound, with the empirical formula C20H23N1, show up the existence of twenty carbon atoms out of which fourteen are aromatic-carbon atoms. These fourteen atoms are well identified in the aromatic region of experimental spectrum 124.531-145.642 ppm. The simulation data also identified the aromatic carbon atoms in the range of 129.662-152.669 ppm. Both the methyl carbon atoms (20C and 21C) located on the spectrum at the position of 42.278 ppm experimentally, while simulation data showed the occurrence of 20C and 21C at 47.822 and 41.068ppm. The methylene carbon atoms (2C, 3C, 10C and 15C) are identified at 24.425, 31.137, 33.054 and 56.498 ppm. Meanwhile, in the spectrum obtained after the DFT calculations, methylene carbon atoms (2C, 3C, 10C and 15C) are detected at 37.477, 34.847, 31.798 and 60.895 ppm.
Vibrational Studies (IR)
The spectra obtained after the experimental [47] and DFT calculations are presented in Figure 1. The prominent vibrations from the different functional groups of the compound have been compared. The material used for the experimental run was salt i.e., amitriptyline hydrochloride, which is showing a broad peak about 3300-3400 cm-1, which might be due to the presence of moisture in the sample material, so the same peak is not observing in the simulated spectra as this was studied using the structure of pure amitriptyline. As there are methyl and methylene groups in the molecule due to which C-H stretching peaks are observed in the range of 3000-2850 cm-1. C-N stretching peaks for the typical tertiary amine functional group observed at about 1250 cm-1 in experimental spectrum while in simulated spectrum C-N stretching observed at 1200 cm-1. The difference might also be due to different environment around the nitrogen atom i.e., amitriptyline hydrochloride was used for experimental observation and amitriptyline was used for DFT calculations. The C=C stretching peaks for aromatic group pointed out near 1600 cm-1 in experimental as well as DFT calculated spectra. Similarly, aromatic C-H stretching and bending are observed in their respective regions Figure 1.
UV-visible spectroscopic analysis
We used the time-dependent density functional theory (TD-DFT), IEFPCM model to look at the electronic spectrum of AMT in gas, water, MeOH, and DMSO (Figure 1). We conducted the calculations using the B3LYP/6-31++G(d,p) level of theory, utilizing the optimized geometry of the ground state. The absorption spectra were experimentally analysed at room temperature in water with a concentration of 5×10−5 M by our group [48]. We measured the highest wavelength in the experiment at room temperature to be 239 nm. The results from Figure 1 showed that the molecule displayed a strong intensity band at 281, 281, 282, and 281 nm in all four solvents. This could be due to the electronic transition of n→π*.
Thermodynamics properties
We calculated the AMT molecule's thermodynamic properties (Gibb's free energy, enthalpy, and entropy) using the following equations.
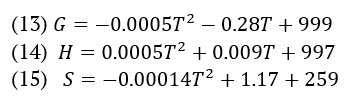
Figure 2 visually illustrates the relationship between the thermodynamic process and temperature. We used temperature to establish parallel equations suitable for linear and quadratic equations.
Molecular Docking Study
We used molecular docking to gain deeper insights into the AMT molecule. We commonly use this method to identify bioactive compounds for drug development by assessing the ligand binding strength to the protein. This ligand interacts with the selected protein using online drug target prediction, such as Swiss ADME-Target prediction. The binding affinity for the two proteins obtained is –8.9 and –9.6 kcal/mol. The low binding energy value demonstrates its bioactive nature. The hydrogen bond distance provides information on the compatibility of the ligand with the proteins.
Figures & Tables
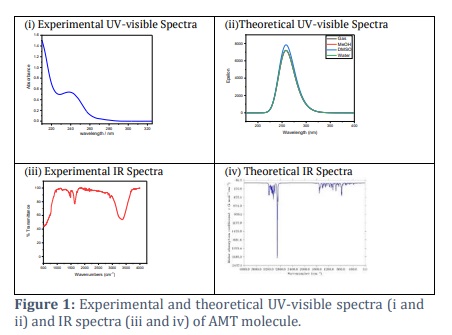
We compared AMT's bond lengths and angles computed by the DFT study with the found experimental values. The average divergence of the discovered bond lengths, which ranged from 0.01 Å to 0.03 Å, agreed with the experimental values. The presence of hydrogen bonding inside molecules may cause the inconsistencies. The results show that the B3LYP function, which uses 6-311++ G(d,p) basis sets, gives the most accurate results close to the experimental values. The B3LYP functional and the 6-311++ G (d, p) basis set were used to find the MEP. Some of the most significant information regarding the hydrogen bonding interactions that occur within the molecules, as well as the potential positively charged and negatively charged sites, is depicted in Figure 1. It was decided to position the region with a higher negative charge on the N atom of the thiadiazole ring, shown in red in Figure 1. Because there are N-H-N hydrogen bonding surfaces close to each other, this spot was chosen because it was thought to be perfect for an electrophilic attack with a V(r) of -0.039 a.u.
Table 1 demonstrates that the AMT has a low value of chemical softness (0.19 ev), so theoretically, it can be considered a nontoxic molecule. Identification of the chemical behaviour of the compound was made possible through the utilization of the electronegativity factor. One of the reasons why a molecule's electronegativity is higher than average is because of the chemical reactivity of the molecule. The compound AMT has a dipole moment (μ) of approximately -1.17 D along the x-axis (μx), -0.08 D along the y-axis (μy), and 0.29 D along the z-axis (μz). Examining the hyperpolarizability value of AMT allows for exploring its potential to investigate novel nonlinear optical (NLO) features. Figure 2 displayed the ELF (Electron Localization Function). The molecule exhibits a covalent bonding region between the C-C and C-N atoms, characterized by the highest LOL value and the highest degree of ELF. The blue ring region corresponds to a large atomic nucleus's valence and inner shell. Figure 2 displays the Electron Localization Function (ELF) of the hydrogen bonding area, represented as a shaded surface map with a projection effect.
HNMR study describes that the protons appeared in their respective regions experimentally or in the DFT case. The variation in the positions of protons for experimental and simulated values might be due to the solvent effect (D2O used in experimental observation while CH2Cl2 for DFT calculations). The 13CNMR data sets showed a good correlation among both calculations; the slight difference might be due to the difference of solvent used for NMR measurements. While searching in literature to provide a comparative NMR spectroscopic results we found a reference in which NMR of amitriptyline as free base and salt have been reported in d6-DMSO [49]. The data is very interesting, and values agree with each other with the minor difference. The six methyl protons are detected at 2.64 ppm in d6-DMSO for hydrochloric acid salt of amitriptyline and when d2-D2O was used as solvent these protons appeared at 2.474. These six protons occupied the position at 2.0 ppm in d6-DMSO when amitriptyline as free base was under study, experimentally. The simulation data indicated that in dichloromethane as deuterated solvent the six protons of methyl groups have appeared in the range of 1.133-2.309 ppm. When we compare the availability of methylene hydrogen atoms in all these four experiments, it is observed that the ranges of appearing the peaks for the subjected protons are very much close to each other. In addition to these methyl and methylene hydrogen atoms, a comparative expected appearance for aromatic hydrogen atom-H26 noted and data sets are well aligned. The peak for H26 hydrogen atom appeared at 5.82 ppm in d6-DMSO while the experiment was taken place as AMP-chloride salt and the presence of H26 was detected at 5.8 ppm when as pure free base was dissolved in d6-DMSO for experiment. In the present study amitriptyline chloride salt was dissolved in d2-D2O for H-NMR study and the peak for H26 atom was detected at 5.55 ppm. In the simulation data we have selected the dichloromethane and pure free base for a comparative study so the peak for H26 appeared at 6.16 ppm. Vibrational (IR) data depicts that the experimental spectrum is nicely correlate with calculated spectrum. The only thing that was different between the calculated and measured data was the C-N stretching peaks for the common tertiary amine functional group. These peaks were seen at about 1250 cm-1 in the measured spectrum and at 1200 cm-1 in the simulated spectrum. Different environments around the nitrogen atom, such as using amitriptyline hydrochloride for experimental observation and amitriptyline for DFT calculations, could potentially explain the difference. As temperature increases, Gibbs free energy (G) decreases, while the values of enthalpy (H) and entropy (S) thermodynamic functions increase, as shown in Figure 2. The same pattern was also observed by Fatima et al. [50]. As temperature escalates, molecule vibrational intensities and translational and rotational energy augment, hence enhancing thermodynamic functions. The thermodynamic properties exhibited fitting factors (R2) of 0.999 for Gibbs free energy, entropy, and enthalpy, respectively. The thermodynamic parameters were calculated by analysing the vibrations of the system using the B3LYP/6-31++G(d,p) method. Gibbs free energy quantifies thermodynamic characteristics of biological systems, such as protein stability, DNA behaviour, and enzyme kinetics. As the temperature increases, the intensity of molecular vibrations and the energy associated with translation and rotation also increase, leading to an increase in thermodynamic functions [51]. Molecular docking is a commonly utilized technique in drug discovery for exploring the relationship between structure and activity, along with biological activity. Molecular docking serves as an effective technique for drug development within the pharmaceutical industry, as it minimizes both the time and cost of synthesis while enhancing the efficacy of pharmaceuticals. People regard it as a modern approach that accurately identifies the precise location where the ligand binds to the protein receptor molecule. We dock the AMT molecule with the two protein receptors (PDB codes: 4IB4 and 8IRV). 4IB4 belongs to chimeric protein. Chimeric, or fusion proteins, are proteins formed by the amalgamation of two or more genes that initially encoded distinct proteins. The translation of this fusion gene produces one or more polypeptides that possess functional characteristics derived from each of the constituent proteins. The 8IVR protein is a guanine nucleotide-binding protein (G-protein). G-proteins act as molecular switches inside cells and help send signals from outside the cell to inside it. The binding affinity for two proteins obtained are –8.9 and –9.6 kcal/mol. Abdulhameed Odhar et al., reported the energy of binding (–7.1 and –7.2 Kcal/mol) for salbutamol against monoamine oxidase [52]. The low binding energy value demonstrates its bioactive nature. The hydrogen bond distance provides information on the compatibility of the ligand with the proteins.
The primary use of Amitriptyline (AMT), a tricyclic antidepressant drug, is to treat depression. This study aims to forecast quantum computational studies using the DFT and molecular docking method. Based on the obtained data, we can conclude:
The study found that the geometrical parameters (bond lengths, angles, and torsion angles) calculated by DFT were very close to the experimental data, with only a difference of 0.01Å to 0.03Å on average. MEP (Molecular Electrostatic Potential) and ELF (Electron Localization Function), which employ distinct colour codes, determine the reactivity, chemical properties, and sites of attack of the molecule. We also computed the softness, electronegativity, chemical hardness, and electrophilicity index values. The FMO study confirms that compounds are both non-toxic and biologically active. In order to look at the molecule's photochemical, electronic, and structural properties, we compare experimental and computational data from spectroscopy (1HNMR, 13CNMR, FTIR, and UV spectroscopy). The experimental and theoretical FT-IR results provide comparable values for the vibrations of C-H, C-N, and C-C. We determined the maximum wavelength (λmax) in the gas phase to be 281.02 nm. In H2O, the λmax was also 281.02 nm, while in methanol it was 280.96 nm, and in DMSO it was 281.03 nm. We found the experimental λmax in H2O to be 262 nm.
We conducted molecular docking analyses on the 4IB4 and 8IRV proteins. When the molecule docks with proteins, the calculated binding energy is -8.9 kcal/mol for 4IB4 and -9.6 kcal/mol for 8IRV. This suggests that the AMT has potential for further investigation in pharmaceuticals.
Acknowledgements
This Project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under grant no. (GPIP: 1236-961-2024). The authors, therefore, acknowledge with thanks DSR for technical and financial support.
Author Contributions
KAA supervised all computational studies. NA and MNA were involved in studying the design and conducting the project. All authors read and approved the final manuscript. All named authors have read the journal's authorship agreement and have reviewed and approved the manuscript. The authors affirm that the members on the list of authors carried out this work.
The authors declare no conflict of interest in publishing this paper.
![]() References
References
- Bendtsen L, Jensen R. Amitriptyline reduces myofascial tenderness in patients with chronic tension-type headache. Cephalalgia, (2000); 20(6): 603–610.
- Boyce P, Judd F. The Place for the tricyclic antidepressants in the treatment of depression. Australian & New Zealand Journal of Psychiatry, (1999); 33(3): 323–327.
- Tomkins GE, Jackson JL, O’Malley PG, Balden E, Santoro JE. Treatment of chronic headache with antidepressants: a meta-analysis11Disclaimer: The views in this article reflect those of the authors and are not intended to represent in any way those of the US Army or the Department of Defense. American Journal of Medicine, (2001); 111(1): 54–63.
- Rub MA, Azum N, Kumar D, Asiri AM. Interaction of TX-100 and antidepressant imipramine hydrochloride drug mixture: surface tension, 1H NMR, and FT-IR investigation. Gels, (2022); 8(3): 159.
- Rub MA, Asiri AM, Azum N, Khan A, Khan AAP et al. Amphiphilic antidepressant drug amitriptyline hydrochloride under the influence of ionic and nonionic hydrotropes; micellization and phase separation. Journal of Industrial and Engineering Chemistry, (2013); 19(6): 1774–1780.
- Rub MA, Azum N, Kumar D, Arshad MN, Khan A, et al. Investigation of solution behavior of antidepressant imipramine hydrochloride drug and non-ionic surfactant mixture: experimental and theoretical study. Polymers, (2021); 13(22): 4025.
- Lagan G, McLure HA. Review of local anaesthetic agents. Current Anaesthesia and Critical Care, (2004); 15(4–5): 247–254.
- Ivandini TA, Sarada BV, Terashima C, Rao TN, Tryk DA, et al. Electrochemical detection of tricyclic antidepressant drugs by HPLC using highly boron-doped diamond electrodes. Journal of Electroanalytical Chemistry, (2002); 521(1–2): 117–126.
- Ruiz-Angel M. Optimised procedures for the reversed-phase liquid chromatographic analysis of formulations containing tricyclic antidepressants. ournal of Pharmaceutical and Biomedical Analysis, (2003); 32(1): 71–84.
- Joron S, Robert H. Simultaneous determination of antidepressant drugs and metabolites by HPLC Design and validation of a simple and reliable analytical procedure. Biomedical Chromatography, (1994); 8(4): 158–164.
- Williams M, Kowaluk EA, Arneric SP. Emerging molecular approaches to pain therapy. Journal of Medicinal Chemistry, (1999); 42(9): 1481–1500.
- Eisenach JC, Gebhart GF. Intrathecal amitriptyline acts as an N-Methyl-D-Aspartate receptor antagonist in the presence of inflammatory hyperalgesia in rats. Anesthesiology, (1995); 83(5): 1046–1054.
- Sawynok J, Reid AR, Esser MJ. Peripheral antinociceptive action of amitriptyline in the rat formalin test: involvement of adenosine. Pain, (1999); 80(1): 45–55.
- Mourik TV, Bühl M, Gaigeot MP. Density functional theory across chemistry, physics and biology. Philosophical Transactions of the Royal Society A. Mathematical, Physical and Engineering Sciences, (2014); 37: 1-5.
- Bochevarov AD, Watson MA, Greenwood JR, Philipp DM. Multiconformation, density functional theory-based pKa prediction in application to large, flexible organic molecules with diverse functional groups. Journal of Chemical Theory and Computation, (2016); 12(12): 6001–6019.
- Mutailipu M, Xie Z, Su X, Zhang M, Wang Y, et al. Chemical cosubstitution-oriented design of rare-earth borates as potential ultraviolet nonlinear optical materials. Journal of the American Chemical Society, (2017); 139(50): 18397–18405.
- Kerru N, Gummidi L, Bhaskaruni SVHS, Maddila SN, Singh P et al. A comparison between observed and DFT calculations on structure of 5-(4-chlorophenyl)-2-amino-1,3,4-thiadiazole. Scientific Reports, (2019); 9(1): 19280.
- Sharma AK, Sameera WMC, Jin M, Adak L, Okuzono C, et al. DFT and AFIR Study on the mechanism and the origin of enantioselectivity in iron-catalyzed cross-coupling reactions. Journal of the American Chemical Society, (2017); 139(45): 16117–16125.
- Yilmazer ND, Korth M. Comparison of molecular mechanics, semi-empirical quantum mechanical, and density functional theory methods for scoring protein–ligand interactions. The Journal of Physical Chemistry B, (2013); 117(27): 8075–8084.
- Domingo L, Ríos-Gutiérrez M, Pérez P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules, (2016); 21(6): 748.
- Joshi R, Pandey N, Yadav SK, Tilak RS, Mishra S, et al. Synthesis, spectroscopic characterization, DFT studies and antifungal activity of (E)-4-amino-5-[N’-(2-nitro-benzylidene)-hydrazino]-2,4-dihydro-[1,2,4]triazole-3-thione. Journal of Molecular Structure, (2018); 1164: 386–403.
- Møller C, Plesset MS. Note on an approximation treatment for many-electron systems. Physical Review, (1934); 46(7): 618–622.
- Gaussian R, Trucks G, Schlegel H, Scuseria G, Robb MA, et al. Gaussian, Inc, Wallingford CT, (2004); 121: 150-166.
- Lu T, Chen F. Multi wfn: a multifunctional wavefunction analyzer. Journal of Computational Chemistry, (2012); 33(5): 580–592.
- Pettersen EF, Goddard TD, Huang CC, Couch GS. UCSF chimera—a visualization system for exploratory research and analysis. Journal of Computational Chemistry, (2004); 25(13): 1605–1612.
- Tirado-Rives J, Jorgensen WL. Performance of B3LYP density functional methods for a large set of organic molecules. Journal of Chemical Theory and Computation, (2008); 4(2): 297–306.
- Momany FA, Appell M, Willett JL, Bosma WB. B3LYP/6-311++G** geometry-optimization study of pentahydrates of α- and β-d-glucopyranose. Carbohydrate Research, (2005); 340(9): 1638–1655.
- Muthu S, Elamurugu Porchelvi E. FTIR, FT-RAMAN, NMR, spectra, normal co-ordinate analysis, NBO, NLO and DFT calculation of N,N-diethyl-4-methylpiperazine-1-carboxamide molecule. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, (2013); 115: 275–286.
- Raajaraman B, Sheela NR, Muthu S. Spectroscopic, quantum computational and molecular docking studies on 1-phenylcyclopentane carboxylic acid. Computational Biology and Chemistry, (2019); 82: 44–56.
- Shanmugavadivu T, Dhandapani M, Vinitha G, Beaula TJ. Quantum chemical calculations, structural, spectral and nonlinear optical investigations of a novel crystal N,N ′-diphenylguanidinium 3,5-dichlorobenzoate. Journal of Physics and Chemistry of Solids, (2019); 130: 69–83.
- Fatima A, Singh M, Singh N, Savita S. Investigations on experimental, theoretical spectroscopic, electronic excitations, molecular docking of Sulfaguanidine (SG): An antibiotic drug. Chemical Physics Letters, (2021); 783: 139049.
- Alaşalvar C, Soylu MS, Ünver H, Ocak İskeleli N, Yildiz M, et al. Crystal structure and DFT calculations of 5-(4-Chlorophenyl)-1-(6-methoxypyridazin-3-yl)-1H-pyrazole-3-carboxylic acid. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, (2014); 132: 555–562.
- Ekennia AC, Osowole AA, Olasunkanmi LO, Onwudiwe DC, Olubiyi O, et al. Synthesis, characterization, DFT calculations and molecular docking studies of metal (II) complexes. Journal of Molecular Structure, (2017); 1150: 279–292.
- Ekennia AC, Osowole AA, Onwudiwe DC, Babahan I, et al. Synthesis, characterization, molecular docking, biological activity and density functional theory studies of novel 1,4‐naphthoquinone derivatives and Pd(II), Ni(II) and Co(II) complexes. Applied organometallic chemistry, (2018); 32(5) e403.
- Huang Y, Rong C, Zhang R, Liu S. Evaluatting frontier energy and HOMO/LUMU gap with descriptors from density functional reactivity theory. J Mol Model, (2017); 23: 3.
- Parr RG, Szentpály L v., Liu S. Electrophilicity Index. Journal of the American Chemical Society, (1999); 121(9): 1922–1924.
- Parr RG, Pearson RG. Absolute hardness: companion parameter to absolute electronegativity. Journal of the American Chemical Society, (1983); 105(26): 7512–7516.
- Parr RG, Donnelly RA, Levy M, Palke WE. Electronegativity: The density functional viewpoint. Journal of Chemical Physics, (1978); 68(8): 3801–3807.
- Chithra S, Mani G, Kumar M, Sevvanthi S, Asif FB, et al. Molecular docking, structural examination, reactive sites identification (Homo–Lumo, Mep) of 6-Phenylpteridine 2, 4, 7-triamine: potential bacterial inhibitor. Analytical Chemistry Letters, (2021); 11(6): 886–898.
- Abraham CS, Prasana JC, Muthu S. Quantum mechanical, spectroscopic and docking studies of 2-Amino-3-bromo-5-nitropyridine by density functional method. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, (2017); 181: 153–163.
- Yang Weitao, Mortier WJ. The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. Journal of the American Chemical Society, (1986); 108(19): 5708–5711.
- Maria Julie M, Prabhu T, Elamuruguporchelvi E, Asif FB, et al. Structural (monomer and dimer), wavefunctional, NCI analysis in aqueous phase, electronic and excited state properties in different solvent atmosphere of 3-{(E)-[(3,4-dichlorophenyl)imino]methyl} benzene-1,2-diol. Journal of Molecular Liquids, (2021); 336: 116335.
- Cassidy C, Halbout JM, Donaldson W, Tang CL. Nonlinear optical properties of urea. Optics Communications, (1979); 29(2): 243–246.
- Savita S, Fatima A, Garima Km, Pooja Km, et al. Experimental spectroscopic, quantum computational, hirshfeld surface and molecular docking studies on 3-Pyridinepropionic acid. Journal of Molecular Structure, (2021); 1243: 130932.
- Al-Muhanna MK, Rub MA, Azum N, Khan SB, et al. Effect of gelatin on micellization and microstructural behavior of amphiphilic amitriptyline hydrochloride drug solution: A detailed study. Journal of Chemical Thermodynamics, (2015); 89: 112–122.
- Rub MA, Khan F, Azum N, Asiri AM, Marwani HM. Micellization phenomena of amphiphilic drug and TX-100 mixtures: Fluorescence, UV-visible and 1H NMR study. Journal of the Taiwan Institute of Chemical Engineers (2016); 60: 32–43.
- Rub MA, Azum N, Alfaifi SYM, Sakib MMH, et al. Assessment of mixed micellization behavior of amitriptyline hydrochloride and TX-165 surfactant mixture in various media (aqueous/NaCl/urea). Journal of Molecular Liquids, (2023); 383: 122073.
- Rub MA, Pulikkal AK, Azum N, Asiri AM. The assembly of amitriptyline hydrochloride + triton X-45 (non-ionic surfactant) mixtures: Effects of simple salt and urea. Journal of Molecular Liquids, (2022); 356: 118997.
- Bhattacharyya PK. NMR spectral study of proton transfer in amitriptyline hydrochloride-chlordiazepoxide hydrochloride combinations in dipolar aprotic solvent. Journal of Pharmaceutical Sciences, (1980); 69: 180-183.
- Fatima A, Bhadoria J, Srivastava SK, Verma I, et al. Exploration of experimental and theoretical properties of 5,5-dimethyl 3-amino-cyclohex-2-en-1-one (amine dimedone) by DFT/TD-DFT with ethanol and DMSO as solvents and molecular docking studies. Journal of Molecular Liquids, (2021); 338: 116551.
- Sajan D, Joseph L, Vijayan N, Karabacak M. Natural bond orbital analysis, electronic structure, non-linear properties and vibrational spectral analysis of l-histidinium bromide monohydrate: A density functional theory. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, (2011); 81(1): 85–98.
- Odhar HA, Hashim AF, Humad SS. Molecular docking analysis and dynamics simulation of salbutamol with the monoamine oxidase B (MAO-B) enzyme. Bioinformation, (2022); 18(3): 304–309.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0![]()

