

Full Length Research Article
In-Silico Perspectives on the Potential Therapeutic Aids of Hesperetin Derivatives for Lung Cancer
Abdulaziz Asiri
Adv. life sci., vol. 11, no. 4, pp. 878-886, November 2024
*– Corresponding Author: Abdulaziz Asiri (amfasiri@ub.edu.sa)
Authors' Affiliations
[Date Received: 28/04/2024; Date Revised: 11/08/2024; Date Published: 15/10/2024]
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Lung carcinoma has become one of the most noteworthy and dangerous health problems discovered today. The disease is mainly caused by smoking and becomes among the leading causes of death spread by growing the cancerous cells into the lining of the lungs and nearby lobes. The FDA has given clearance to numerous drugs and chemotherapy agents; nevertheless, they can be exceedingly costly as well as frequently fall short of entirely addressing the ailment. In the medical management of lung cancer, new medicines or active leads with high efficacy and minimal toxicity are required in this era.
Methods: The study has been conducted with the intent to uncover a prospective approach to treating lung cancer through structural modification of Hesperetin by creating its analogs to enhance its efficacy compared to its parent compound with the computational drug design. The analysis has been conducted with various approaches followed by PASS prediction, ADME, toxicity profile, molecular docking, data filtration, and anticancer activity.
Results: All the compounds showed satisfactory criteria in each parameter that was assessed. The data mining was done carefully by pointing out the compounds that had the greatest value among all the compounds in each investigation, which resulted in a total of 3 compounds out of 50.
Conclusions: Finally, the selected compounds were further analyzed for MD simulation studies. Afterward, PCA analysis was also conducted in order to get the lead compound with this additional investigation.
Keywords: Lung cancer; Anti-cancer lead; Hesperetin; Drug designing; Molecular docking; MD simulation; PCA
Introduction![]()
In the 21st century, the era witnessed and recorded the discovery of around 200 distinct types of cancer, each named for the tissue in which it was originally discovered [1]. Among them, the highly invasive and rapidly spreading cancer is lung cancer in the United States of America (USA) caused by dangerous cells which spread quickly if left untreated and lead to mortality [2]. In 2018, 9.6 million individuals, or one in six fatalities, died from various forms of cancer, according to the World Health Organization’s annual report [3]. As a result of healthcare facility cuts brought on by the fear that COVID-19 contamination may cause a sharp decline graph in cancer deaths [4], followed by an increase in advanced-stage illness and, ultimately, higher mortality due to a lack of access to affordable healthcare, cancer has the second highest incidence of death in the United States and is a major global public health concern. Lung cancer was responsible for more than 2.2 million new cases and roughly 1.8 million fatalities in 2020 [5]. By 2022-23, lung cancer claimed the lives of about 350 Americans every day on average. Projections show that lung cancer kills 2.5 times as many people as colorectal cancer and more than all forms of pancreatic, breast, and prostate cancer combined [6].
Cancerous cells grow abruptly and become uncontrollable leading to a decline in people’s lives. India was considered as the second largest consumer of tobacco which affects the health of a person [7] leaving behind its impact. One of the main leading causes of lung cancer is smoking [8] which leads to genetic mutation through DNA adduct formation. In some people, this cancer may be connected to radon (222Rn) exposure by building materials who never smoked [9]At the very earliest stage, only patients with non-small cell lung cancer in a small fraction got detected [10]. The multiple cellular mechanisms implicated in the etiology of lung cancer deliver promising opportunities for target-specific remedies. The common major pathways targeted included the RTKs pathway, Protein kinase C (PKC), mitogen protein kinase (MAPK) systems, and many more are altered in this disease by oncogenesis through mutation or overexpression triggering cell proliferation. The currently ongoing therapies include chemotherapy, radiation, and surgery for lung cancer today [11]. Chemotherapy and radiotherapy, on the other hand, can induce a variety of adverse effects in patients, and surgical treatment has some dangers and side effects as well [12]. Through all the literature shreds of evidence and demands, it becomes essential to find the alternative for a better cure.
With the demanding trend of getting back to nature, a massive increment in the number of people searching for the active lead compound obtained by nature that not only prevents but successfully treats human ailments. In the view of result, chasing for active natural compounds and vigorously searching the molecular mechanism and biological activity of anti-lung cancer cells, as well as the development of Chinese medications, was quite critical for preventing and treating lung cancer. One of the natural and demanding compounds Hesperetin, a flavanone, derived from the compound hesperidin was focused which can be extracted from various citrus species in considerable quantities [13] With the help of accordance research, the compound was treated and proven to have the benefits of acquiring normal blood pressure, reducing blood vessel fragility, suppressing cancer, anti-virus, and anti-cancer activities [14]. By raising oxidative stress, hesperidin can hasten cancer cell apoptosis which is taken as Hesperetin [15][16]
From extensive research studies and pieces of literature on hesperidin absorption, bioavailability, and pharmacokinetics, the findings illuminated that hesperidin can be taken orally in humans and absorbed only in the form of aglycon called Hesperetin [17]. They are keeping that in mind and previously reported bioactivities, this molecule Hesperetin has been picked up and altered by adding or substituting with different functional groups. Gemcitabine, a well-known drug commonly used to treat lung cancer variants such as small-cell lung cancer was used as drug control in this investigation. In contrast, the aim objectifies to proposed Hesperetin derivatives as a potential anticancer drug candidate via a computer-aided drug designing approach.
Hesperetin (3’,5,7- trihydroxy-4-methoxy flavanone), a metabolite of hesperidin, is a polyphenolic component of citrus fruits. Also, popular as the flavanone subgroup of flavonoids [18]. The IUPAC name of Hesperetin was (2S)-5,7-dihydroxy-2-(3-hydroxy-4-methoxyphenyl)-2,3-dihydrochromen-4-one. The natural compound possesses a huge spectrum of biological activities proven as anti-inflammatory, anti-viral, anti-cancer, antineoplastic, and antioxidative effects [19]. Gaining more attention towards the molecule, it’s also an accessible ingredient as well as cost-effective. Several research studies have also been conducted in vivo and in vitro to examine their effects proving that they may play a promising ingredient.
Methods![]()
Retrieval and preparation of ligands database
The very first step included the retrieval of the compound of CID: 72281 in the canonical SMILE format. Next, the build-up step was to draw the main molecule by uploading the canonical SMILE format as the first step for the online tool MolOpt followed by the second step to choose the bio isosteric replacement method [20]. The rules from the data mining(fast) replacement method were chosen to draw the derivatives. 50 derivatives were drawn (Figure 1) in the smiles format and were converted into PDB format by the ChemDB chemoinformatic portal [21] for further analysis (https://cdb.ics.uci.edu/cgibin/BabelWeb.py).
In silico prediction of activity spectra for substances (PASS)
To evaluate the antifungal, antibacterial, antiviral, and antineoplastic activities, we availed the PASS prediction tool (https://www.way2drug.com/passonline/index.php). Initially, the derivatives structures of Hesperetin were drawn with the help of the MolOpt online tool (https://xundrug.cn/molopt) followed by uploading the smile format of the main chemical compound Hesperetin retrieved by PubChem (https://pubchem.ncbi.nlm.nih.gov). Afterward, the SMILE format of derived structures generated by the MolOpt online tool was uploaded to the PASS prediction tool to interpret the above-mentioned mass spectrum activities. Measuring values were determined by the concepts Pa and Pi, the probability of active and inactive molecules. For a compound to be considered the Pa and Pi values should lie between 0.000 to 1.00, and the Pa value must be greater than Pi.
Target retrieval and preparation
There are numerous proteins available for lung cancer. Among them, the basic and preliminary proteins of lung cancer were selected (PDB ID: 1X2J), (PDB ID: 2P85) and retrieved with the assistance of a protein data bank (https://www.rcsb.org/) in the PDB format. The macromolecule was then prepared by removing water molecules and integrating the hydrogen bonds via the BIOVIA Discovery Studio Visualizer 2021 [22,23]. To this extent, the active sites were taken from the literature and rechecked by the CASTp server (http://sts.bioe.uic.edu/castp/).
Analysis of ADME and drug-likeness profile
The pharmacokinetics properties addressed by ADMET, and profile determination are the most crucial and necessary steps in drug development [24][23]This profile generates the pharmacokinetics activities of the compound to indicate the further step of whether the compounds have properties to become drug-like molecules or not. To follow up on this criterion, the Swiss ADME [25] and pkCSM [26] prediction tools were used.
Molecular Docking: Blind and Target Docking
The molecular docking was performed by PyRx 0.8 version [27]. All the compounds and macromolecules were uploaded and converted into pdbqt format. Whole macromolecules were selected for grid formation in the case of blind docking [28]. In contrast, the active site was selected in the grid formation step in target docking. The results were saved in the .csv file format and were visualized with the help of the PyMol visualization tool [29]. Best compounds were taken out and revalidated with the help of Auto dock 4.2 version followed by the preparation of ligands and protein [30].
Compound’s lethality prediction
To determine and predict acute rat toxicity, side effects parameters were analyzed by online servers GUSAR (http://www.way2drug.com/gusar/acutoxpredict.html) [31], and ADVER-PRED (http://www.way2drug.com/adverpred/) [32] were used.
Anticancer sensitivity prediction
The PacMann’s database (https://huggingface.co/spaces/jannisborn/paccmann) was used to determine the anti-cancer activity of screened compounds against different cancer cell lines [33].
Molecular dynamics determination
Based on the molecular interaction and analysis of docking results, the top hits of 3 compounds were pointed out for MD simulation studies. The simulation was accomplished by the GROMACS (GROningen Machine for Chemical Simulations) package of version 5.1.2. the simulation was conducted at 300 K [34]. For the extraction of the complexes and topologies of the proteins, the GROMACS package was used by gmx grep module. For all the hit compounds, force field CGenFF (CHARMm general force field) was used [35]. Each complex was solvated within a cubic box with a TIP3 (Transferable Intermolecular potential) model. boundary conditions were then applied and neutralized the charges by adding salts. For the minimization, the system was acquired by 25,000,000 steps by steepest descent algorithm. The temperature was then raised while maintaining the constant NVT (Number of particles at constant Volume & constant Temperature) and NPT (Number of particles at constant Pressure & constant Temperature) conditions. Post the equilibration phase, through Ewald method, mesh was applied [36]. The simulation was then conducted at 100 ns to look into the molecular dynamics.
Principal component analysis
Within the Galaxy platform, we implemented the analytical method of Principal Component Analysis (PCA) https://usegalaxy.eu/ to gain insights and investigate the conformational changes of the complex interactions that occurred between protein and derivatives. With the aid of PCA, the conformations were highlighted and identified notably within the 100 ns trajectory of lung proteins and selected derivatives [37].
Results![]()
PASS prediction spectrum analysis
A total of 50 compounds were analyzed and all the results indicated a greater Pa value than Pi. The primary concern of pass prediction was to determine the antineoplastic activity that was found in all the compounds, other activities were also noted such as antibacterial, antifungal, and anti-inflammatory.
Pharmacokinetics features analysis
It has been observed that all derivative compounds successfully satisfy the criteria of ADME. The ADME profile investigation was done by the Swiss ADME and pkCSM online tool. For all the compounds, the bioavailability score was noted down as 0.55 and no BBB (Blood blood-brain barrier) was noted. Interestingly, all the compounds followed Lipinski’s rule as well.
Molecular interaction analysis
The molecular docking of whole proteins was performed with the help of PyRx 0.8 version in which the highest energy against protein 1X2J was -10.3 kcal/mol to -10.2 kcal/mol. Six compounds remained within the docking with range -9.6 kcal/mol, nine remained within the range -9.2 kcal/mol to -9.7 kcal/mol, and seven remained within the range -9.1 kcal/mol to -9.0 kcal/mol and rest showed the energies between ranges -9.0 kcal/mol to -7.8 kcal/mol. On the other hand, the highest energies evaluated for protein 2P85 were noted between the range of -9.4 kcal/mol to -10.4 kcal/mol. The parent compound Hesperetin showed the energies -9.2 kcal/mol and -8.9 kcal/mol for both proteins. The remaining compounds showed energy between the ranges of -8.9 kcal/mol to -7.81 kcal/mol. The highest docked complexes were visualized with the PyMol to check whether the compound perfectly binds at the desired site. Out of 50 compounds, 3 compounds were picked up with analogs 12, 16, & 17 from proteins, analog 12, 17 from protein 1, and analog 16 from protein 2 with the best binding site and energies for further analysis. However, most of the compounds showed the best result from the original parent compound Hesperetin in both types of docking. The control Hesperetin was noted with a binding energy of -8 kcal/mol in protein 2P85, and -5.1 kcal/mol for the protein 1X2J which was lower than the derived compounds. The active residues for protein 1X2J were noted down as ILE 421, ASP 422, HIS 424, TYR 426, ASN 442, and PRO 492, while for protein 2P85, only 1 active residue was there i.e., ASN 227 (Figure 2A-D).
The data depicted better results than the parent compound with the highest binding affinity of -9.5kcal/mol. After the analysis of the protein-ligand interaction of proteins 2P85 and 1X2J, the interacting residues of hydrogen bonding, Pi- alkyl, alkyl, van der Waals and amide were discussed with the diagram in (Figure 2E and F). Further analysis of molecular dynamic studies will be investigated for these compounds.
Toxicity profiling
The selected compound was analyzed and observed that no compounds showed carcinogenicity and also showed a non-toxic AMES profile. Additionally, all the compounds showed the non-carcinogenic activity successfully predicted.
Further acute toxicity was conducted to determine the compound’s lethality by determining the side effects that may be caused. This assessment was done by GUSAR and Adver-Pred databases individually. By GUSAR database prediction, it was analyzed that Hesperetin, and all the analogs were non-toxic. Hesperetin was categorized as Class 5 while the selected analogs were categorized as Class I. Another database found no unusual effect.
Cell line studies
For the prediction of anticancer activity, PaccMann database was used using the structure-activity relationship algorithm. The analogs were inserted to test against a variety of cell lines to see the anti-cancer properties in a broad-spectrum area. The output result was measured in two different datasets CCLE and GDSC. Cancer Cell Line Encyclopedia (CCLE) provides a collection of human cancer cell lines from various tumors. While Genomics of Drug Sensitivity in Cancer (GDSC) assessed the drug sensitivity of cell lines. The maximum effectivity was demonstrated by analog 17 as in cell line NCIH209 = IC50 2.952, SBC5 = 2.275, NCI196 = 2.685, 908462 = 3.801, 907173 = 3.241, 1240141 = 3.583 followed by the dataset CCLE & GDSC which were better than control. Interestingly, all the derived analogs or compounds have depicted better results than the parent compound.
Appraising the stability of docked complex: MD Simulation
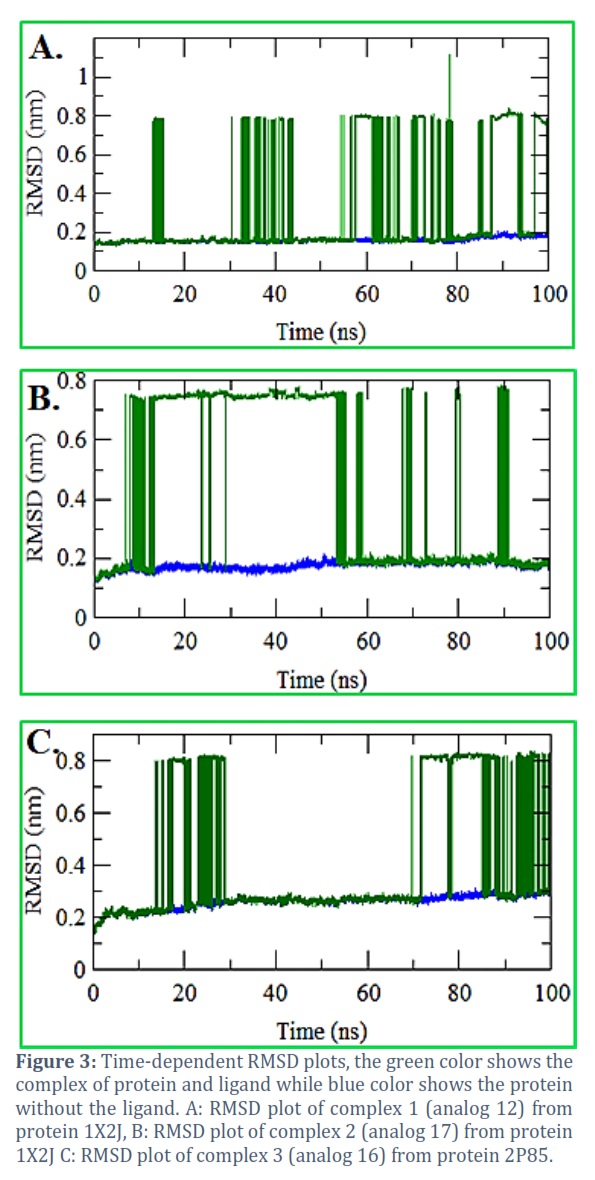
To outline the stability of complexes of top hits from docking compounds, the MD simulation was run at 100 ns using the GROMACS software. To carefully examine the stability of the desired complexes within the ligands and complexes, the graphs of RMSD were plotted.
Root-mean-square deviation (RMSD) analysis
RMSD depicts the steadiness of protein, the miniature of RMSD, the greater the stability. In the steady flow inspection, the RMSD was analyzed for the protein backbone and ligand. The RMSD of top hits analog 12 as compound 1 and analog 17 as compound 2 were noted as 0.78 nm and 0.18 nm for protein 1, while the analog 16 as compound 3 from protein 2 was noted as 0.29 nm respectively (Figure 3). From all the plots, it was stated that compound 2 from protein 1 i.e., 1X2J found more effective having the minimum deviations and stronger stability than the rest two compounds.
Principal Component Analysis
The principal component analysis was put to use and investigated the trajectories of a docked complex for better analysis and comparison of three analogs of Hesperetin with both the lung cancer proteins. The fraction of diversity defined by PCI for compound 1 was noted as 10.1%, 9.4% for compound 2, and 17.2% for compound 3 indicating the trajectories linked with each protein. In the PCA plots, it was stated that the lower the variance proportion, the greater the stability. In light of the phenomenon, compound 2 from protein 1 found greater stability depicting a lower variance. The graphical representation of the plot is presented in Figure 4.
Figures & Tables
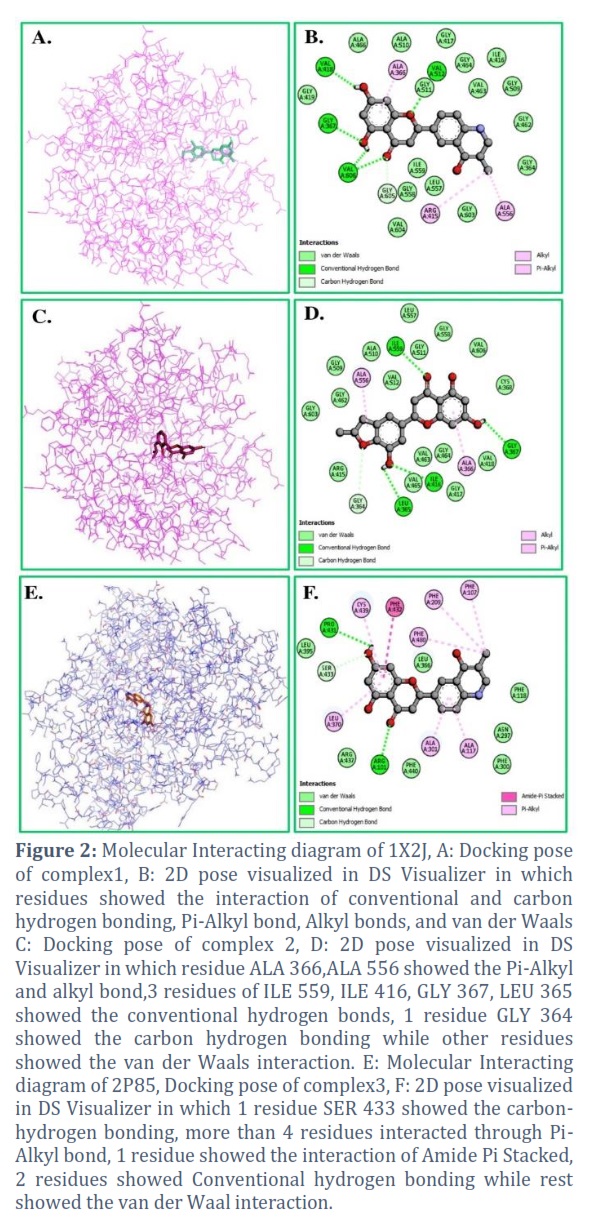

The analysis of PASS prediction was performed with the online tool way2drug. All the derived chemical structures showed anti-viral, antibacterial, antifungal, and antineoplastic effectiveness. The parameter is widely used to assess the probability of active and inactive compounds [38]. The ADMET profile represents the absorption, desorption, metabolism, excretion, and toxicity properties that play a vital role in drug design [39]. The derivative compounds successfully meet the expectations of Lipinski’s five rules as well [40]. Moreover, for target site docking, the active residues of proteins were taken from the literature and also verified from CASTp 3.0 tool [41]. ASN 227 was the lone active residue found in protein 2P85, whereas ILE 421, ASP 422, HIS 424, TYR 426, ASN 442, and PRO 492 were listed as the active residues for protein 1X2J [46, 47]. The ADME properties were already determined and proven to the desired satisfactory result by the Swiss ADME tool. The validation of ADMET determination was done again by the pkCSM tool toxicity profiling [21]. A quantitative investigation into lung cancer has been carried out using derivatives of Hesperetin. In order to develop new inhibitors, a molecular modeling method was implemented [42].
The investigation through PASS Prediction suggested that most of the compounds derived from Hesperetin could convey anti-cancer properties. For more efficacy evaluation, the targeted docking and auto-docking interaction were used to calculate the binding affinity against lung cancer [1]. The compounds that showed notable binding affinities went for further investigation such as pharmacokinetics features, toxicity profiling, anti-cancer cell activity, MD simulation studies, and PCA Analysis. After careful investigation, the study demonstrated that compound 2 i.e., analog 17 from protein 1X2J showed promising lead candidate properties. The analog showed a greater binding affinity with no toxicity than the Hesperetin. It also revealed the anti-cancer activity and maximum efficacy in the cell line among the top 3 hits. The RMSD noted for this compound was 0.18 nm and 9.4% lower variance by PCA. Therefore, by these attributes, it can be concluded that this analog can be treated as the lead candidate for further development and optimization. Additionally, this compound offers promising drug development prospects against lung cancer by giving its significant properties. Further investigation can build on this candidate to develop effective natural therapeutics to overcome this ailment.
Acknowledgments
The author is thankful to the Deanship of Graduate Studies and Scientific Research at University of Bisha for supporting this work through the Fast-Track Research Support Program.
The author declare that there is no conflict of interest regarding the publication of this paper.
![]() References
References
- Cui W, Aouidate A, Wang S, Yu Q, Li Y, et al. Discovering Anti-Cancer Drugs via Computational Methods. Frontiers in Pharmacology, (2020); 11: 733.
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, et al. Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians, (2021); 71 (3): 209-249.
- Schmitt FC, Bubendorf L, Canberk S, Chandra A, Cree IA, et al. The World Health Organization Reporting System for Lung Cytopathology. Acta Cytologica, (2023); 67 (1): 80-91.
- Dela Cruz CS, Tanoue LT, Matthay RA. Lung Cancer: Epidemiology, Etiology, and Prevention. Clinics in Chest Medicine, (2011); 32 (4): 605-644.
- Carrillo-Perez F, Morales JC, Castillo-Secilla D, Molina-Castro Y, Guillén A, et al. Non-small-cell lung cancer classification via RNA-Seq and histology imaging probability fusion. BMC Bioinformatics, (2021); 22(1):454.
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA: A Cancer Journal for Clinicians, (2022); 72 (1): 7-33.
- Singh N, Agrawal S, Jiwnani S, Khosla D, Malik PS, et al. Lung Cancer in India. Journal of Thoracic Oncology, (2021); 16 (8): 1250-1266.
- Collin J. Tobacco control, global health policy and development: towards policy coherence in global governance. Tobacco Control, (2012); 21(2): 274-280.
- Rudin CM, Avila-Tang E, Samet JM. Lung Cancer in Never Smokers: A Call to Action. Clinical Cancer Research (2009); 15 (8): 5622-5625.
- Guo Q, Liu L, Chen Z, Fan Y, Zhou Y, et al. Current treatments for non-small cell lung cancer. Frontiers in Oncology, (2022); 12:945102.
- Debela DT, Muzazu SG, Heraro KD, Ndalama MT, Mesele BW, et al. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Medicine, (2021); 9: 205031212110343.
- Khan T, Date A, Chawda H, Patel K. Polysaccharides as potential anticancer agents-A review of their progress. Carbohydrate Polymers, (2019); 210: 412-428.
- Ramteke, Prerna Yadav, Umesh CS. Hesperetin, a Citrus bioflavonoid, prevents IL-1β-induced inflammation and cell proliferation in lung epithelial A549 cells. NISCAIR-CSIR, India, (2019); 57: 7-14.
- Muhammad T, Ikram M, Ullah R, Rehman S, Kim M. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-κB Signaling. Nutrients, (2019); 11(3): 648.
- Wang Y, Liu S, Dong W, Qu X, Huang C, Yan T, Du J. Combination of hesperetin and platinum enhances anticancer effect on lung adenocarcinoma. Biomedicine & Pharmacotherapy, (2019); 113: 108779.
- Jiao Q, Xu L, Jiang L, Jiang Y, Zhang J, Liu B. Metabolism study of hesperetin and hesperidin in rats by UHPLC-LTQ-Orbitrap MS n . Xenobiotica, (2020); 50 (11): 1311-1322.
- Ávila-Gálvez MÁ, Giménez-Bastida JA, González-Sarrías A, Espín JC. New Insights into the Metabolism of the Flavanones Eriocitrin and Hesperidin: A Comparative Human Pharmacokinetic Study Antioxidants, (2021); 10 (3): 435.
- Alipour M, Sharifi S, Samiei M, Shahi S, Aghazadeh M, et al. Synthesis, characterization, and evaluation of Hesperetin nanocrystals for regenerative dentistry. Scientific Report, (2023); 13 (1): 2076.
- Alipour M, Pouya B, Aghazadeh Z, SamadiKafil H, Ghorbani M, et al. The Antimicrobial, Antioxidative, and Anti-Inflammatory Effects of Polycaprolactone/Gelatin Scaffolds Containing Chrysin for Regenerative Endodontic Purposes. Stem Cells International, (2021); 1-11: :3828777.
- Modee R, Mehta S, Laghuvarapu S, Priyakumar UD. MolOpt: Autonomous Molecular Geometry Optimization Using Multiagent Reinforcement Learning. Journal of Physical Chemistry B, (2023); 127 (48): 10295-10303.
- Chen J, Swamidass SJ, Dou Y, Bruand J, Baldi P. ChemDB: a public database of small molecules and related chemoinformatics resources. Bioinformatics (2005); 21 (22): 4133-4139.
- Sarkar M, Nath A, Kumer A, Mallik C, Akhter F, et al. Synthesis, molecular docking screening, ADMET and dynamics studies of synthesized 4-(4-Methoxyphenyl)-8-Methyl-3, 4, 5, 6, 7, 8-Hexahydroquinazolin-2 (1H)-one and quinazolinone derivatives. Journal of Molecular Structure, (2021); 1244.
- Singh R, Bhardwaj VK, Sharma J, Das P, Rituraj R. Discovery and in silico evaluation of aminoarylbenzosuberene molecules as novel checkpoint kinase 1 inhibitor determinants. Genomics, (2021); 113: 707-715.
- Al Azzam K. SwissADME and pkCSM Webservers Predictors: an integrated Online Platform for Accurate and Comprehensive Predictions for In Silico ADME/T Properties of Artemisinin and its Derivatives. Kompleksnoe Ispolʹzovanie Mineralʹnogo syrʹâ/Complex Use of Mineral Resources/Mineraldik Shikisattardy Keshendi Paidalanu, (2023); 325: 14-21.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Report, (2017); 7: 42717.
- Pires DE V, Blundell TL, Ascher DB. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. The Journal of Medicinal Chemistry, (2015); 58 (9): 4066-4072.
- Mai C, NA, P.I. Thai Journal of Pharmaceutical Sciences. Thai Journal of Pharmaceutical Sciences, (2018); 93-97.
- Nath A, Kumer A, Zaben F, Khan Md W. Investigating the binding affinity, molecular dynamics, and ADMET properties of 2,3-dihydrobenzofuran derivatives as an inhibitor of fungi, bacteria, and virus protein. Beni-Suef University Journal of Basic and Applied Sciences, (2021); 10: 36.
- Schrodinger L, DeLano W. PyMol. (2020); http://www.pymol.org/pymol, version = {2.4.0}.
- Morris GM, Huey R, Lindstrom W, Michel F, Belew RK, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry, (2009); 30 (16): 2785-2791.
- Lagunin A, Zakharov A, Filimonov D, Poroikov V. QSAR Modelling of Rat Acute Toxicity on the Basis of PASS Prediction. Molecular Informatics, (2011); 30 (2-3): 241-250.
- Ivanov SM, Lagunin AA, Rudik AV, Filimonov DA, Poroikov VV. ADVERPred-Web Service for Prediction of Adverse Effects of Drugs. Journal of Chemical Information and Modeling, (2018); 58 (1): 8-11.
- Cadow J, Born J, Manica M, Oskooei A, Rodríguez Martínez M. PaccMann: a web service for interpretable anticancer compound sensitivity prediction. Nucleic Acids Research, (2020); 48 (W1): W502-W508.
- Stenberg S, Stenqvist B. An Exact Ewald Summation Method in Theory and Practice. The Journal of Physical Chemistry A, (2020); 124 (19): 3943-6.
- Fischer NM, Van Maaren PJ, Ditz JC, Yildirim A, Van Der Spoel D. Properties of Organic Liquids when Simulated with Long-Range Lennard-Jones Interactions. Journal of Chemical Theory and Computation, (2015); 11 (7): 2938-2944.
- Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics, (2006); 22 (21): 2695-2696.
- Khan MKA, Ahmad S, Rabbani G, Shahab U, Khan MS. Target‐based virtual screening, computational multiscoring docking and molecular dynamics simulation of small molecules as promising drug candidate affecting kinesin‐like protein KIFC1. Cell Biochemistry & Function, (2022); 40 (5): 451-472.
- Islam S, Hosen MA, Ahmad S, ul Qamar MT, Dey S, et al. Synthesis, antimicrobial, anticancer activities, PASS prediction, molecular docking, molecular dynamics and pharmacokinetic studies of designed methyl α-D-glucopyranoside esters. Journal of Molecular Structure, (2022); 1260: 132761.
- Vardhan S, Sahoo SK. In silico ADMET and molecular docking study on searching potential inhibitors from limonoids and triterpenoids for COVID-19. Computers in Biology and Medicine, (2020); 124: 103936.
- Hanee U, Rahman MR, Matin and MM. Synthesis, PASS, In Silico ADMET and thermodynamic studies of some galactopyranoside esters. Physical Chemistry Research, (2021); 9(4): 591-603.
- Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Research, (2018); 46: W363-W367.
- Sulimov VB, Gribkova IV, Kochugaeva MP, Katkova EV, et al. Application of Molecular Modeling to Development of New Factor Xa Inhibitors. Biomed Research International, (2015); 2015: 1-15.
- GA, Adeniji SE. Binding profile of protein–ligand inhibitor complex and structure based design of new potent compounds via computer-aided virtual screening. Journal of Clinical Tuberculosis and Other Mycobacterial Diseases, (2021); 24: 100256.
![]()