

Review Article
Neimann-Pick Diseases: Beyond Lipid Accumulation – Genetic, Diagnostics, and Therapeutic Strategies
Hind M Naffadi
Adv. life sci., vol. 11, no. 4, pp. 703-713, November 2024
*- Corresponding Authors: Hind M Naffadi (hmnaffadi@uqu.edu.sa)
Authors' Affiliations
[Date Received: 21/02/2024; Date Revised: 18/08/2024; Date Available Online: 15/10/2024]
Abstract![]()
Introduction
Methods
Discussion
Conclusion
References
Abstract
Rare genetic disorders are the group of disorders/diseases that occur in such a low count that they are not considered much active market for therapeutics unless encouraged by appropriate incentives and support. They are too rare to be fully investigated and managed by health professionals. Genetic disorders are caused by change in genes and are often serious and complex. Around the world, most affected individuals are found to be children. They can be progressive and might get worse as children grow older. Niemann-Pick diseases (NPD) is one such group of rare genetic diseases that are characterized by unwanted and abnormal accumulation of lipids within the cells. In this review, we discuss the history, genetic basis, clinical manifestations and diagnostic approaches for NPD. We further discuss the pathophysiology of accumulation of lipids and their impact on cellular functions and organ systems and various management strategies.
Keywords: Rare disease; Genetic mutation; Lipid metabolism; Cholesterol trafficking; Neurological abnormalities
Introduction![]()
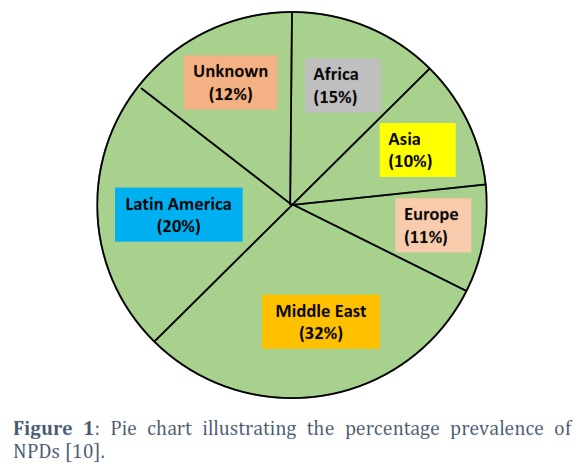
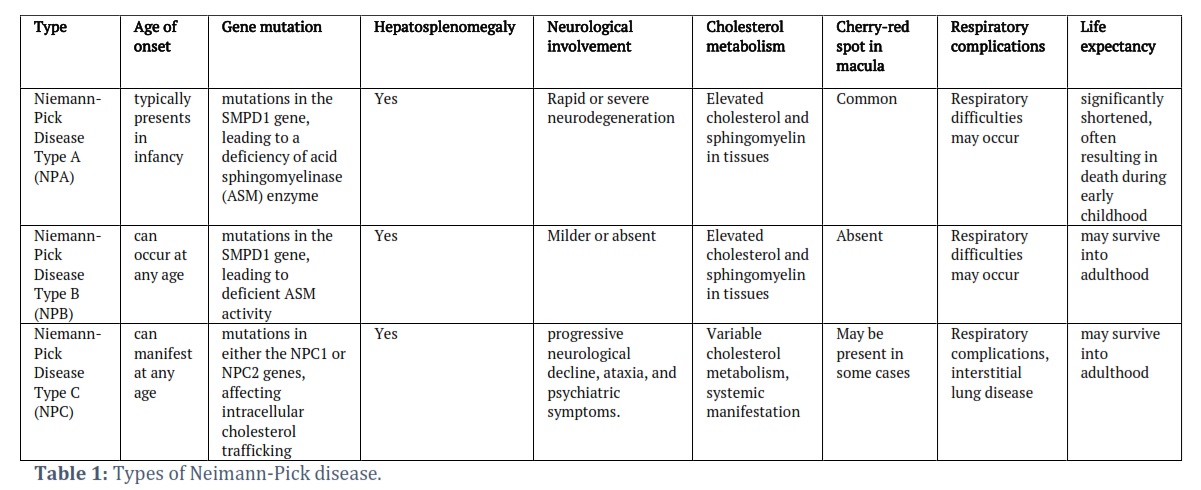
There are certain groups of rare genetic disorders that disrupt the lysosomal storage inside the human body resulting in disrupted molecular and cellular mechanisms. Niemann-Pick disease (NPD) is one example of such disorder that is caused by mutations in genes crucial for processing and transport of lipids (sphingomyelin and cholesterol) [1,2]. Lipid metabolism is severely affected in all three types of NPD [3,4]. It fatally affects various tissues and organs leading to progressive degeneration of crucial organs of the body [5,6]. There are three types of Neiman-Pick disease [7] (Table 1). Niemann-Pick type A (NPA) is caused by a deficiency of acid sphingomyelinase [8]. It is present in early infancy with rapid disease progression and characterized by hepatosplenomegaly, developmental delays, and a shortened lifespan. Type B (NPB) is also caused by a deficiency of the same enzyme, and it can occur in either childhood or later in life. NPB often exhibits milder symptoms affecting mainly visceral function such as hepatosplenomegaly. In hepatosplenomegaly, both liver and spleen get enlarged beyond their normal sizes. On the other hand, NPC (type C of NPD) is caused by the mutations in the genes that are crucial to cholesterol trafficking. NPC1 and NPC2 are the primarily affected genes in this case [9]. NPC constitutes a wide range of symptoms including neurological abnormalities, liver dysfunction and splenomegaly as well in some cases. It is the most dangerous type of NPD as it can show anytime from infancy to adulthood. It is quite a challenging task to diagnose NPD due to their rarity and complexity in clinical context [6]. Based on a recent study of 602 patients over 15 years, global distribution and prevalence of NPC is shown in Figure 1 [10].
However, the rarity of NPDs contributes to the diagnostic challenges [11]. Clinical presentations may mimic other more common conditions necessitating specialized testing such as genetic analysis and lipid profiling for the accurate diagnosis. As of now, therapeutic interventions remain limited, often focusing on symptomatic management. Effective treatment is hindered by the complexity of the underlying molecular mechanisms and the rarity of these conditions. Moreover, the rarity of these diseases seeks a need for increased awareness, innovative research and collaborative efforts.
Research in the NPD field has significantly progressed after the researchers could elucidate the genetic basis and molecular mechanism involved in lipid metabolism, especially sphingomyelin and cholesterol processing. Further, the specific gene mutations were identified which are linked to each subtype, paving the way for refined diagnostic methods and emerging therapeutic strategies. The devastating factor about this rare disease is that it manifests itself as multi-systemic disorders affecting vital organs of the body including liver, spleen and nervous system.
NPD bring about life -altering consequences for affected individuals and their families. The progression of symptoms, especially in the severe forms, can significantly compromise the quality of life, leading to physical abilities, cognitive decline and emotional distress. NPD follows an autosomal recessive inheritance pattern, meaning that both parents must carry a mutated gene for the disorder to manifest in their child [12]. Due to this reason, parents often become apprehensive of family planning emphasizing the importance of genetic counseling. Further, the smaller patient population poses challenges to advocacy and fund-raising efforts.
The review aims to provide a holistic and up-to-date understanding of NPD, contributing to the ongoing dialogue in research, clinical practice, and public awareness. The multifaceted approach encompasses genetic, clinical, and psychosocial dimensions, fostering a comprehensive resource for healthcare professionals, researchers, policymakers, and the broader community.
Methods
![]()
Literature Search and Selection Criteria
Google scholar, Pubmed, Scifinder and google web were used to obtain data for this review paper. Various keywords were used to retrieve the required information from different published research and review articles. Published papers consulted for this review were taken from year 2017-2024.
Discussion![]()
Genetic basis of Niemann-Pick disease and pathophysiology
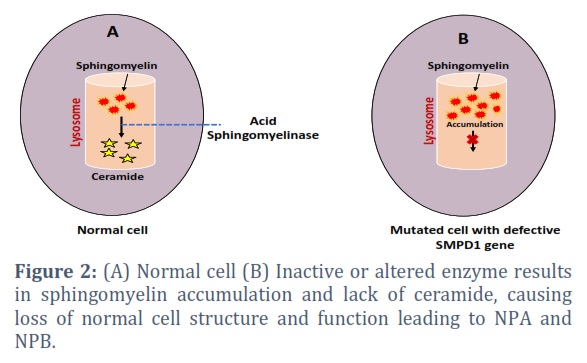
Niemann-Pick diseases (NPD) are a group of rare inherited metabolic disorders characterized by the abnormal accumulation of lipids, particularly sphingolipids, within various tissues [13]. There are three primary types of Niemann-Pick disease: Type A, Type B, Type C. Each type has distinct genetic and clinical characteristics, and they are caused by mutations in different genes. Niemann-Pick Type A (NPA) is a rare and severe genetic disorder characterized by a deficiency of acid sphingomyelinase (ASM) enzyme activity [14]. NPA is primarily caused by mutations in the SMPD1 gene, located on chromosome 11p15.1 (Figure 2) [15]. This particular gene encodes ASM which plays a crucial role in lipid metabolism by hydrolyzing sphingomyelin into ceramide and phosphocholine [16]. That ceramide is involved in many cellular processes including cell death, differentiation, apoptosis, inflammation and senescence. Various types of mutations are responsible for affecting the ASM’s activity. Among them, missense mutation is a common type in NPA that involves single-nucleotide substitutions leading to amino acid changes in ASM enzyme [15], impairing its catalytic activity and affecting its ability to breakdown sphingomyelin. Nonsense mutations, another type of mutation, introduce premature stop codons in the SMPD1 gene that results in a non-functional ASM. Nonsense mutations often lead to a more severe form of NPD type A. A deficiency of normal ASM can also occur due to genetic mutations in NPA blocking normal degradation of sphingomyelin. Undegraded sphingomyelin then accumulates within lysosomes disrupting the normal turnover of cellular components and resulting in the overstuffed lysosomes and the formation of foam cells.
The presence of lipid-laden foam cells contributes to tissue damage, particularly in organs with high lipid turnover such as the liver, spleen and lungs. The molecular consequences extend to lysosomal dysfunction, impacting cellular homeostasis and contributing to the characteristic lipid storage seen in Niemann-Pick diseases. Specific mutations in the SPMD1 gene contribute to a spectrum severity in NPA [17]. Certain mutations may result in a more severe phenotype with an earlier onset and rapid disease progression, while others may lead to a milder form of disorder.
NPB is primarily associated with mutations in the SMPD1 gene, which is located on chromosome 11p15.1 [18]. In individuals with NPB, mutations in the SMPD1 gene results in a deficiency or dysfunction of ASM enzyme. The genetic basis of NPB exhibits heterogeneity with different mutations leading to variations in the severity of the disorder, age of onset and the extent of organ involvement.
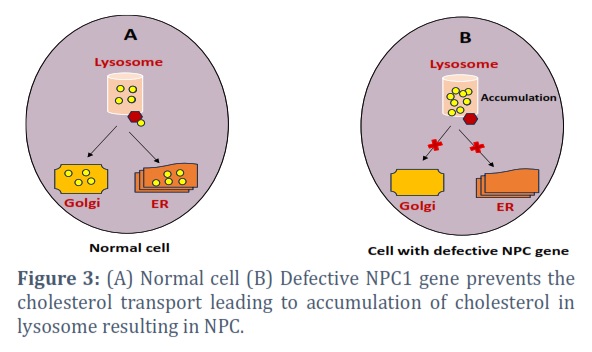
NPC is distinguished by distinguished by the buildup of cholesterol and glycosphingolipids within the late endocytic pathway [19]. It occurs primarily due to mutations in two genes- NPC1 and NPC2. The diverse spectrum of mutations including missense, nonsense, and splice site mutations, influences the severity and age of onset of NPC. These genetic mutations result in defective cholesterol metabolism causing the hallmark lysosomal lipid storage bodies. The NPC1 gene is located on chromosome 18q11-q12 and it encodes a transmembrane protein crucial for intracellular cholesterol transport, while the NPC2 gene, located on chromosome 14q24.3 and encodes a soluble lysosomal protein involved in lipid metabolism [20, 21]. Mutations in NPC1 disrupt the process and cause the abnormal accumulation of cholesterol within lysosomes forming characteristic storage bodies (Figure 3). Similarly, mutations in NPC2 contribute to impaired cholesterol trafficking and lysosomal storage causing altered endocytic pathways. Cholesterol accumulation in endolysosomal system also impacts autophagy- a crucial cellular process for recycling damaged cellular components. Moreover, Cholesterol is vital for cellular membranes, and its abnormal accumulation in Niemann-Pick disease type C affects membrane fluidity and signalling, contributing to neurodegeneration. Lipid accumulation disrupts cellular communication and signalling pathways. Sphingolipids like ceramide and sphingosine-1-phosphate are crucial for cell signalling, and their dysregulation in Niemann-Pick diseases interferes with pathways involved in apoptosis, cell survival, and inflammation [22]. This disruption likely contributes to neurodegeneration in Niemann-Pick disease type C and other systemic issues in types A and B.
Impact on neurological functions
The impact of NPDs on neurological function is a central and often devastating aspect of these disorders. NPA an NPB are characterized by a rapid and severe neurological deterioration. The accumulation of sphingomyelin in neurons leads to progressive damage causing ataxia, muscle weakness and cognitive decline. Children affected by NPA typically experience a regression of developmental milestones and fail to achieve normal neurological maturation [23]. The severity of neurological involvement contributes significantly to the limited life expectancy in this case. On the other hand, NPC presents with a more varied and protracted neurological course. The effect on neurological function is multifaceted and include ataxia, dysarthria, dysphagia and cognitive decline [24]. VSGP, a specific oculomotor abnormality is a characteristic feature. Seizures and psychiatric symptoms such as mood disorders and psychosis may also occur. The central nervous system’s vulnerability to lipid accumulation is particularly evident in NPDs. The accumulation of sphingolipids and cholesterol within neurons impact membrane integrity, intracellular trafficking and cellular signalling pathways. These alterations contribute to the degeneration of neurons and the progressive loss of neurological functions. The cerebellum, brainstem and other regions crucial for motor coordination and cognitive processes are particularly affected leading to the observed neurological symptoms.
Advancements in understanding enzymatic deficiencies – targeted interventions
NPDs are characterized by enzymatic deficiencies that results in the abnormal accumulation of lipids within cells. Mutations disrupt cholesterol egress from lysosomes, leading to its abnormal accumulation which can affect multiple organs. Advancements in understanding the enzymatic deficiencies of Niemann-Pick diseases have paved the way for therapeutic exploration. Enzyme replacement theory (ERT) involves replacing the deficient or malfunctioning enzyme, acid sphingomyelinase, in individuals with Niemann-Pick disease. ERT involves the production of therapeutic enzymes primarily in mammalian cells, with some also produced in plant and yeast cells. These enzymes undergo extensive processing to ensure their activity, high bioavailability and lesser susceptibility to the degradation. Despite the success of ERT, however, several factors pose challenges to its efficacy enhancement. These challenges involve optimizing the interaction of the enzymes with cell membranes and improving their internalization into target cells, reducing the immunogenic response to the enzymes and overcoming the blood brain barrier when targeting neuronal cells [25]. Nevertheless, ERT stands a highly successful example of targeted biologic therapies, proved to be both effective and safe in treating various genetic rare diseases, saving the lives of newborns and significantly improving the quality of patients’ life. Recent research has focused on developing ERT with olipudase alfa, a recombinant human ASM to treat NPA and NPB. One study evaluated the effects of ERT on lipid parameters and inflammatory markers in five adults with NPA over 26 weeks. The patient showed higher levels of triglycerides, PCSK9, apoB48, oxidized LDL, and TNF-α, and lower levels of HDL-C and apoA1 compared to a reference group. ERT led to improvements in total cholesterol, triglycerides, LDL-C, small dense LDL-C, oxidized LDL, and apoB100. Despite a slight reduction in HDL-C, apoA1 levels increased, and TNF-α levels decreased. The study suggests that ERT improves dyslipidemia in NPD-B patients, potentially through reductions in TNF-α and PCSK9 [26]. The interplay of enzymes and lipid metabolism in these disorders remains a subject of ongoing investigation, offering hope for future therapeutic advancements.
Moreover, NPDs share overlapping clinical features with various metabolic and neurodegenerative disorders making the differential diagnosis challenging. Conditions such as Gaucher disease and Tay-Sachs disease exhibit similarities in terms of sphingolipid accumulation [27], but specific enzyme assays and genetic testing help distinguish each type. Additionally, mucopolysaccharidoses (MPS), Wilson disease, and mitochondrial disorders may present with hepatomegaly, neurological symptoms or systematic involvement requiring specific biochemical and genetic tests for accurate differentiation [28]. Rett syndrome, characterized by neurological regression also shares features with NPDs but specific clinical criteria and genetic testing for MECP2 mutations aid in distinguishing the two [29].
Diagnostic approaches for NPD
Genetic testing
Genetic testing plays a significant role in the diagnosis and management of NPD. Since these disorders are quite heterogenous genetically, identifying the specific genetic mutations is essential for accurate diagnosis and classification into type A, B and C of Niemann-Pick diseases [30]. DNA sequencing and mutation analysis are the common methods employed to identify pathogenic variants in genes SPMD1, NPC1 and NPC2 [31]. Molecular genetic testing is quite helpful in identification of specific mutations as it not only confirms the diagnosis but also give insight into the potential severity of the disease. Its further aids in prognostication process. For instance, certain mutations in NPC1 or NPC2 genes may be associated with a severe or milder form of NPC [32]. Genetic testing is also significant for carrier identifications in families helpful for family planning decisions and genetic counselling [33]. Further advancements in genetic testing technologies such as next-generation sequencing (NGS), targeted gene panels, whole exome sequencing and microarrays have facilitated more comprehensive screening of affected genes [34, 35]. One study has discovered and assessed the pathogenicity of three mutations which were previously unidentified on one known mutation in NPC1 gene through whole exome sequencing [36]. Such studies are helpful for the genetic and prenatal diagnosis of the disease.
Moreover, as research progresses and new genetic variations linked to these conditions are uncovered, the accuracy and precision of genetic testing methods are being enhanced. This advancement plays a role, in improving how these disorders are clinically managed and in exploring treatment options. With the development of medicine genetic testing will continue to play a central role in the comprehensive approach to comprehending diagnosing and addressing NPD.
Biochemical testing
Biochemical testing plays a role, in assessing individuals suspected of having NPD. One important test involves checking sphingomyelin levels in tissues in blood samples [37]. High sphingomyelin levels indicate issues with sphingomyelinase activity which are common in NPA and NPB. Enzyme tests that measure acid sphingomyelinase activity, are useful for confirming these subtypes diagnosis. For example, a study showed that lower acid sphingomyelinase activity in leukocytes and higher plasma 7 ketocholesterol levels were closely linked to the disease’s onset and severity [38]. In the case of NPC, testing typically includes examining oxysterols like cholestane 3β,5α,6β triol, which can build up due to cholesterol trafficking problems [39]. Increased levels of oxysterols in blood or cerebrospinal fluid may indicate NPC presence. Additionally using filipin staining on fibroblasts is a method that reveals abnormal cholesterol storage patterns specific, to NPC.
Imaging modalities
In individuals with NPC, magnetic resonance imaging (MRI) is often used to detect brain abnormalities because it provides detailed images of the nervous system. A recent study found that patients with NPC had a thalamus volume and higher quantitative susceptibility mapping in the pulvinar nuclei compared to the control group [40]. A correlation was also noted with the help of MRI between increased quantitative susceptibility mapping in the pulvinar nuclei clusters which was associated with severe disease symptoms. MRI imaging technique support the idea that iron levels play a significant role in neurodegeneration process in NPC [41].
There are several other functional imaging techniques that are used to study metabolic and physiological changes associated with NPD [42]. Examples include positron emission tomography and photon emission computed tomography. These methods are helpful in deep analysis of how the disease affects the brain function. Another method is ultrasound that is used to check a clinical sign of NPD i.e. hepatosplenomegaly, in which organ size can be monitored [43]. Fundoscopy is another imaging method that is used for eye examination in case of cherry red spot which is a clinical feature in certain type of NPD [44]. Despite all the imaging modalities, diagnosis of NPD has many challenges. Even among the affected individuals of the same family, a range of clinical symptoms can be observed within each subtype [45].
Approaches for management and treatment of NPD
Symptomatic management
The treatment of NPD symptoms plays a role, in caring for patients given the range of symptoms that impact different parts of the body [46]. A collaborative approach is taken to manage the signs and enhance the quality of life for those affected. Liver and spleen enlargement are common in NPA and NPB. Managing symptoms involves monitoring organ sizes and providing support to ease associated issues. Regular imaging and assessments help evaluate the effectiveness of treatments like enzyme replacement therapy or substrate reduction therapy which aim to reduce buildup in organs. NPC often shows signs like ataxia, dysarthria and cognitive decline. Managing these symptoms requires a team effort involving neurologists, physical therapists and occupational therapists. Treatments focus on preserving mobility improving communication skills and offering assistance. New pharmacological methods like miglustat aim to slow down decline and enhance function [47]. Certain forms of NPD, type C can lead to breathing problems. Symptomatic care includes support measures such as assisted ventilation when needed. Regular monitoring of breathing function along with intervention is crucial to manage issues like pneumonia and ensure lung health [48]. NPC may also present symptoms such, as mood disorders and psychosis [49].
Treating these symptoms involves teamwork, between brain specialists and mental health experts. Using medications and therapies that offer support can help ease health issues and enhance the quality of life for those affected. The distinctive cherry red spot seen in the eye’s macula might appear in forms of NPD. Although it acts as a clue its effect on eyesight is usually minor. Routine eye checkups are done to keep an eye, on eye health and deal with any vision related worries.
Enzyme Replacement Therapy
Enzyme Replacement Therapy (ERT) has shown promise as a treatment approach, for managing aspects of NPD, particularly type A and B [50]. Acid sphingomyelinase plays a crucial role in breaking down sphingomyelin and when it’s lacking this lipid accumulates in lysosomes. ERT entails administering acid sphingomyelinase to supplement the enzyme function and aid in breaking down the accumulated sphingomyelin. The impact of ERT on NPD type A and B has been most notable in addressing hepatosplenomegaly, a symptom [51]. By reducing the sphingomyelin stored in the liver and spleen ERT helps diminish organ enlargement and alleviate related symptoms. Significant enhancements in organ enlargement have been observed in studies indicating that ERT is effective in slowing disease progression enhancing quality of life and increasing survival rates for those affected [52]. However, challenges remain when it comes to tackling the symptoms of NPD through ERT. While enzyme replacement therapy has proven effective in animal studies by reducing sphingomyelin accumulation in the system overcoming the blood brain barrier to ensure sufficient enzyme penetration remains a hurdle [53]. The limited spread of enzymes, to the brain complicates the management of issues that often significantly influence patient prognosis and quality of life.
Substrate Reduction Therapy (SRT)
SRT is a treatment strategy that aims to lessen the effects of NPA and NPB by decreasing the buildup of sphingomyelin. It works by inhibiting the production of sphingolipids which reduces the amount, for storage and slows down disease progression [54]. The use of SRT has been particularly effective in managing hepatosplenomegaly in Niemann Pick diseases A and B where it helps reduce the accumulation of sphingomyelin in the liver and spleen. Clinical studies have shown that SRT can improve organ enlargement and related symptoms leading to a quality of life for those affected. This treatment method involves taking an administered molecule that blocks the first step in glycosphingolipid synthesis. The goal is to lower the production rate of glycosphingolipids to counteract issues with their breakdown thereby restoring a balance between their creation and breakdown rates. Ketogenic dietary therapies (KDT) have been successful in managing seizures in patients with metabolic disorders including those with seizure disorders. Since individuals with Niemann Pick type C often face drug seizures and nerve cell degeneration KDT could offer advantages. For instance, there’s a documented case involving a 17-year girl, with NPC who received both miglustat medication and KDT over three years [26].
Despite the patient’s neurodegeneration, there was a decrease, in seizure occurrences fewer hospital visits due to worsened seizures and an improvement in alertness. This indicates that KDT could be a beneficial treatment option for individuals with NPC. It has demonstrated effectiveness in reducing seizure frequency decreasing medication needs and offering neuroprotection for those with epilepsy and IMD. However, due to concerns about cholesterol metabolism in NPC patients it is important to be cautious, about their intake of lipids [55].
Emerging therapy and clinical trial cases
The field of NPD is changing as new treatments are being explored and people are actively taking part in studies. Rare metabolic conditions, like types A, B and C have always been difficult to treat. Recent progress in understanding how they work has opened up possibilities for treatment. One exciting option is gene therapy, which aims to fix the underlying issues by giving working copies of the genes that are missing [56, 57]. Researchers are currently studying gene therapy methods, in animals and small clinical trials to see how safe and effective they are with the hope that this could lead to a treatment that offers lasting benefits [58].
Challenges include ensuring effective delivery of therapeutic genes to target cells, addressing immune responses, and optimizing vector design. Small molecule therapies, including pharmacological chaperones and modulators, are being explored for their potential to correct enzyme function or enhance the cellular response to the underlying metabolic defects [59]. These compounds aim to restore proper cellular function and reduce the accumulation of lipid substrates. Several small molecule therapies are currently in preclinical and clinical development, with ongoing trials assessing their safety and efficacy. Researchers are actively working on optimizing SRT approaches, aiming to enhance drug delivery to the central nervous system. Strategies include developing more brain-penetrant compounds and exploring alternative administration routes. SRT, which has shown efficacy in addressing hepatosplenomegaly, remains an essential component of emerging therapeutic strategies, especially for types A and B.
ERT continues to be a focus of research, with efforts to improve enzyme formulations, increase efficacy, and enhance targeting. Novel ERT approaches seek to overcome challenges related to blood-brain barrier penetration, aiming to provide more comprehensive treatment for neurological manifestations, particularly in NPC. Novel nanotechnology-driven enzymes replacement strategies have been explored for such lysosomal storage disorders [53]. The emerging trend in Niemann-Pick disease research involves exploring combination therapies that target multiple aspects of the disease simultaneously [61]. Combining different modalities, such as gene therapy with small molecule treatments or ERT with SRT, aims to synergistically address the complexities of organ involvement and improve overall outcomes [62, 63]. Genetic testing can comprise specific examination of NPC1/NPC2 genes or specialized gene panels, and it can be complemented by conducting enzymatic assay tests for acidic sphingomyelinase and evaluating the biomarker N-palmitoyl-O-phosphocholine serine (PPCS). Study has suggested that beyond its role in variant classification, the biomarker PPCS could potentially function as an indicator of the severity or progression of the disease [10].
Identification of biomarkers
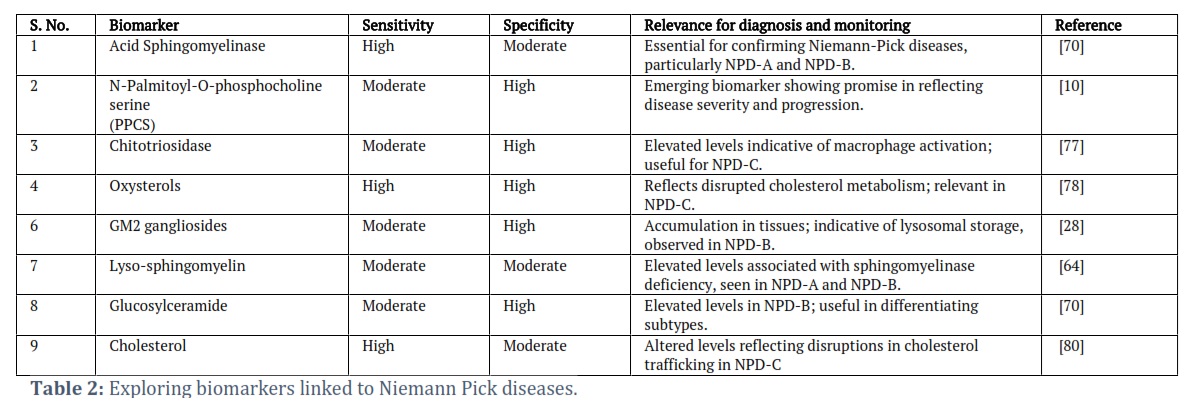
The identification of reliable biomarkers for the diagnosis and prognosis of NPDs has become a crucial focus in advancing clinical management and therapeutic interventions [64]. Biomarkers play a pivotal role in facilitating early and accurate diagnosis, enabling timely interventions and improved patient outcomes (Table 2) [65]. Several potential biomarkers associated with various aspects of NPDs are under investigation, reflecting the complex and heterogeneous nature of these disorders. Genetic testing remains a cornerstone in the diagnosis of Niemann-Pick diseases, providing definitive confirmation of the underlying genetic mutations associated with each subtype [64, 66]. Advanced technologies such as NGS, has paved the way for the identification of particular pathogenic variants, enhancing the accuracy and efficiency of genetic testing [67]. Genetic biomarkers not only aid in diagnostic certainty but also contribute to family planning decisions and genetic counselling. Assessing biochemical markers associated with sphingolipid metabolism is fundamental in diagnosing and monitoring NPDs [68]. Elevated levels of sphingomyelin and other sphingolipids in blood or tissues serve as indicative biomarkers. Furthermore, the analysis of oxysterols, which’re byproducts of cholesterol breakdown has become a potential tool, for diagnosis especially in NPC. Different imaging techniques like MRI offer indicators for evaluating organ involvement and disease progression [69. Hepatosplenomegaly, cerebellar atrophy and other structural irregularities detected through imaging play a role in the process and continuous monitoring. The presence of a cherry spot in the macula is an observation in certain types of NPDs and acts as a significant indicator during eye examinations. Recognizing this sign helps with diagnosis. Provides insights into the overall impact of the disease on the body. Studies have also examined blood markers related to processes in NPC1 patients [71]. Optical coherence tomography (OCT) presents an opportunity to study retinal changes in systemic neurological disorders affecting both adults and children [72]. These OCT characteristics, such as. Spots on the retina, SND (subfoveal neuroretinal detachment) changes in retinal layer thicknesses and alterations in inner and outer retinal layer integrity serve as indicators, for neuroinflammation or neurodegeneration. For example, OCT angiography was utilized to assess deposit patterns in layers of NPB patients underlining the importance of combining imaging techniques for identifying deposit patterns and diagnosing NPA patients [73].
The evaluation of these OCT biomarkers plays a role, in monitoring the status of the nervous system allowing for a comprehensive approach necessary for accurate diagnosis and effective management of these conditions. Researchers are increasingly exploring biomarkers found in cerebrospinal fluid (CSF) to understand their potential in reflecting involvement of the nervous system [74]. Analysis of CSF, which includes studying proteins or metabolites can provide insights into the neurodegenerative aspects of Niemann Pick diseases aiding both in diagnosis and prognosis. Recent research has validated proteins in CSF from NPC1 patients that warrant further investigation [75]. Additionally, a recent study highlights the use of neurofilament light chain in CSF as a novel biomarker for evaluating both the severity of symptoms and treatment response in NPC1 [76]. As new therapies advance through research and early clinical trials there is optimism within the Niemann Pick community regarding potentially transformative treatments. However, challenges such as selection, long term safety concerns and addressing the nature of these diseases continue to persist. Ongoing collaboration and active participation in trials are crucial for propelling these therapies towards wider availability and improved outcomes, for patients.
Tables and Figures
In summary, this detailed review provides an examination of the rare genetic condition, NPD. It discusses background, genetic basis, clinical features and current research findings in the field of NPD research. In such condition, early detection through enzyme and genetic testing is crucial to prevent the premature mortality. There is always a chance that healthcare professionals miss or delay the proper diagnosis due to the overlapping symptoms, owing to its rarity and diversity. The present study helps understand the complexity, disease management, psychological, social and economic aspects in dealing with NPD.
The author(s) declare no conflict of interest.
![]() References
References
- Vanier MT. Niemann-pick diseases. The Handbook of Clinical Neurology, (2013); 113: 1717-1721.
- Kolodny EH. Niemann-Pick disease. Current Opinion in Hematology, (2000); 7(1): 48-52.
- Percival BC, Gibson M, Wilson PB, Platt FM, and Grootveld M. Metabolomic studies of lipid storage disorders, with special reference to Niemann-Pick type C disease: A critical review with future perspectives. International Journal of Molecular Sciences, (2020); 21(7): 2533.
- Rodrigues KF, Yong WTL, Bhuiyan MSA, Siddiquee S, Shah MD, and Venmathi Maran BA. Current understanding on the genetic basis of key metabolic disorders: a review. Biology, (2022); 11(9): 1308.
- Vitiello L, De Bernardo M, Nuzio SG, Mandato C, Rosa N, Vajro P. Pediatric liver diseases and ocular changes: What hepatologists and ophthalmologists should know and share with each other. Digestive and liver disease, (2020); 52(1): 1-8.
- Toledano-Zaragoza A, and Ledesma MD. Addressing neurodegeneration in lysosomal storage disorders: Advances in Niemann Pick diseases. Neuropharmacology, (2020); 171: 107851.
- Schuchman EH, Desnick RJ. Types a and B Niemann-pick disease. Molecular Genetics and Metabolism, (2017); 120(1-2): 27-33.
- Pfrieger FW. The Niemann-Pick type diseases-A synopsis of inborn errors in sphingolipid and cholesterol metabolism. Progress in Lipid Research, (2023); 90:101225.
- Vanier, MT. Niemann-Pick disease type C. Orphanet Journal of Rare Diseases, (2010) 5: 1-18.
- Guatibonza Moreno P, Pardo LM, Pereira C, Schroeder S, Vagiri D, et al. At a glance: the largest Niemann-Pick type C1 cohort with 602 patients diagnosed over 15 years. European Journal of Human Genetics, (2023); 31(10): 1108-1116.
- Hendriksz CJ, Anheim M, Bauer P, Bonnot O, Chakrapani A, et al. The hidden Niemann-Pick type C patient: clinical niches for a rare inherited metabolic disease. Current Medical Research and Opinion, (2017). 33(5): 877-890.
- Patterson M. Niemann-Pick disease type C. University of Washington, Seattle, (2020); 1993-2024.
- McGovern MM, Avetisyan R, Sanson B-J, and Lidove O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Current Medical Research and Opinion, (2017); 12(1): 1-13.
- Pinto C, Sousa D, Ghilas V, Dardis A, Scarpa M, and Macedo MF Acid sphingomyelinase deficiency: a clinical and immunological perspective. International Journal of Molecular Sciences, (2021); 22(23): 12870.
- Zampieri S, Filocamo M, Pianta A, Lualdi S, Gort L, Coll MJ, Sinnott R, Geberhiwot T, Bembi B, Dardis A. SMPD1 mutation update: database and comprehensive analysis of published and novel variants. Human Mutation, (2016) 37(2): 139-147.
- Deshpande D, Gupta SK, Sarma AS, Ranganath P, Jain S JMN, et al. Functional characterization of novel variants in SMPD1 in Indian patients with acid sphingomyelinase deficiency. Human Mutation, (2021); 42(10):1336-1350.
- Chetta M, Guacci A, Rizzo F, Marchese G, Operto FF, et al. Infantile spasms in early-onset Niemann–Pick disease with a novel compound heterozygous mutations in SMPD1 gene. Elsevier, (2015); 2(6-7): 155-158.
- Mukherjee SB, Pandey M, Kapoor S, and Priya TP. Infant with type A Niemann Pick disease and undetectable Niemann Pick cells in bone marrow. Indian Pediatrics, (2012); 49(6): 490.
- Reddy JV, Ganley IG, and Pfeffer SR. Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PLoS One, (2006); 1(1): p. e19.
- Bernardo A, De Nuccio C, Visentin S, Martire A, Minghetti L, et al. Myelin Defects in Niemann-Pick Type C Disease: Mechanisms and Possible Therapeutic Perspectives. International Journal of Molecular Sciences, (2021); 22(16): 8858.
- Bulut FD, Bozbulut NE, Özalp Ö, Dalgiç B, Mungan NÖ, et al. Diagnostic value of plasma lysosphingolipids levels in a Niemann-Pick disease type C patient with transient neonatal cholestasis. Journal of Pediatric Endocrinology and Metabolism, (2022); 35(5): 681-685.
- Newton J, Palladino EN, Weigel C, Maceyka M, Gräler MH, et al. Targeting defective sphingosine kinase 1 in Niemann–Pick type C disease with an activator mitigates cholesterol accumulation. Journal of Biological Chemistry, (2020); 295(27): 9121-9133.
- Djafar JV, Johnson AM, Elvidge KL, and Farrar MA. Childhood dementia: A collective clinical approach to advance therapeutic development and care. Pediatric Neurology, (2023); 139: 76-85.
- Cariati I, Masuelli L, Bei R, Tancredi V, Frank C, and D’Arcangelo G. Neurodegeneration in niemann–pick type c disease: An updated review on pharmacological and non-pharmacological approaches to counteract brain and cognitive impairment. International Journal of Molecular Sciences, (2021); 22(12): 6600.
- Marchetti M, Faggiano S, & Mozzarelli A. Enzyme replacement therapy for genetic disorders associated with enzyme deficiency. Current Medicinal Chemistry, (2022); 29(3), 489-525.
- Garside B, Ho JH, Kwok S, Liu Y, Dhage S, et al. Changes in PCSK 9 and apolipoprotein B100 in Niemann–Pick disease after enzyme replacement therapy with olipudase alfa. Orphanet Journal of Rare Diseases, (2021); 27;16(1):107.
- Ryckman AE, Brockhausen I, and Walia, JS. Metabolism of glycosphingolipids and their role in the pathophysiology of lysosomal storage disorders. nternational Journal of Molecular Sciences, (2020); 21(18): 6881.
- Zakir F, Mohapatra S, Farooq U, Mirza MA, and Iqbal Z, Introduction to metabolic disorders. Drug Delivery Systems for Metabolic Disorders, (2022); Elsevier.1-20.
- Djafar JV, Johnson AM, Elvidge KL, and Farrar MA. Childhood dementia: A collective clinical approach to advance therapeutic development and care. Pediatric Neurology, (2023); 139: 76-85.
- Kannan P, Nanda Kumar MP, Rathinam N, Kumar DT, Ramasamy M. Elucidating the mutational impact in causing Niemann–Pick disease type C: an in silico approach. Journal of Biomolecular Structure and Dynamics, (2023); 41(17): 8561-8570.
- Dardis A, Zampieri S, Gellera C, Carrozzo R, Cattarossi S, et al. Molecular genetics of Niemann-Pick Type C disease in Italy: an update on 105 patients and description of 18 NPC1 novel variants. Journal of Clinical Medicine, (2020); 9(3): 679.
- Xu Y, Zhang Q, Tan L, Xie X, and Zhao Y. The characteristics and biological significance of NPC2: Mutation and disease. Mutation Research – Reviews in Mutation Research, (2019); 782: 108284.
- Metcalfe SA. Genetic counselling, patient education, and informed decision-making in the genomic era. Seminars in Fetal and Neonatal Medicine, (2018); 23(2):142-149.
- Kumar K, Cowley M, Wetterstrand K, Watson J, Crick F, Sanger F, Nicklen S, et al. Next-generation sequencing and emerging technologies. in Seminars in thrombosis and hemostasis. (2019); Thieme Medical Publishers 333 Seventh Avenue, New York, NY 10001, USA.
- Sudrié-Arnaud B, Snanoudj S, Dabaj I, Dranguet H, Abily-Donval L, et al. Next-Generation Molecular Investigations in Lysosomal Diseases: Clinical Integration of a Comprehensive Targeted Panel. Diagnostics, (2021); 11(2): 294.
- Guan C, Gan X, Yang C, Yi M, Zhang Y, and Liu S. Whole-exome sequencing analysis to identify novel potential pathogenetic NPC1 mutations in two Chinese families with Niemann–Pick disease type C. Neurological Sciences, (2022); 43(6): 3957-3966.
- Sitarska D, and Ługowska AJ. Laboratory diagnosis of the Niemann-Pick type C disease: an inherited neurodegenerative disorder of cholesterol metabolism. Metabolic Brain Disease, (2019); 34 (5): 1253-1260.
- Hu J, Maegawa GH, Zhan X, Gao X, Wang Y, et al. Clinical, biochemical, and genotype‐phenotype correlations of 118 patients with Niemann‐Pick disease Types A/B. Human Mutation, (2021); 42(5): 614-625.
- Cooper J, Church H, Wu HY. Cholestane-3β, 5α, 6β-triol: further insights into the performance of this oxysterol in diagnosis of Niemann-Pick disease type C. Molecular Genetics and Metabolism, (2020); 130(1): 77-86.
- Ravanfar P, Syeda W, Rushmore R, Moffat B, Lyall A, et al. Investigation of Brain Iron in Niemann-Pick Type C: A 7T Quantitative Susceptibility Mapping Study. AJNR – American Journal of Neuroradiology, (2023); 44(7): 768-775.
- Yang F, Guan Y, Feng X, Rolfs A, Schlüter H, and Luo J. Proteomics of the corpus callosum to identify novel factors involved in hypomyelinated Niemann-Pick Type C disease mice. Molecular Brain, (2019); 12(1): 1-11.
- Amin MM. An Introduction, in Handbook of Research on Critical Examinations of Neurodegenerative Disorders. IGI Global, (2019); 195-216.
- Masi B, Perles-Barbacaru TA, Bernard M, and Viola A. Clinical and preclinical imaging of hepatosplenic schistosomiasis. Trends in Parasitology, (2020); 36(2): 206-226.
- Padungkiatsagul T, Leung L-S, Moss HE. Retinal diseases that can masquerade as neurological causes of vision loss. Current Neurology and Neuroscience Reports, (2020); 20 (11): 51.
- Encarnação M, Ribeiro I, David H, Coutinho MF, Quelhas D, et al. Challenges in the Definitive Diagnosis of Niemann–Pick Type C-Leaky Variants and Alternative Transcripts. Genes, (2023); 14(11): 1990.
- Lawson J, Harrell E, Deruiter J, Pathak S, Pondugula S, et al. Genetic disease and Niemann-Pick disorders: novel treatments and drug delivery systems. Drug Delivery Systems for Metabolic Disorders, Elsevier, (2022); 161-175.
- Pineda M, Walterfang M, and Patterson MC. Miglustat in Niemann-Pick disease type C patients: a review. Orphanet Journal of Rare Diseases, (2018); 13(1): 1-21.
- Bonnot O, Klünemann H-H, Velten C, Martin JVT, and Walterfang M. Systematic review of psychiatric signs in Niemann-Pick disease type C. The World Journal of Biological Psychiatry, (2019); 20(4):320-332.
- Rego T, Farrand S, Goh AM, Eratne D, Kelso W, et al. Psychiatric and cognitive symptoms associated with Niemann-Pick type C disease: neurobiology and management. CNS Drugs, (2019); 33 (2): 125-142.
- Li M. Enzyme replacement therapy: a review and its role in treating lysosomal storage diseases. Pediatric Annals, (2018); 47(5): e191-e197.
- Marchetti M, Faggiano S, and Mozzarelli A. Enzyme replacement therapy for genetic disorders associated with enzyme deficiency. Current Medicinal Chemistry, (2022); 29(3): 489-525.
- Jones SA, McGovern M, Lidove O, Giugliani R, Mistry PK, et al. Clinical relevance of endpoints in clinical trials for acid sphingomyelinase deficiency enzyme replacement therapy. Molecular Genetics and Metabolism, (2020); 131(1-2): 116-123.
- Del Grosso A, Parlanti G, Mezzena R, and Cecchini M. Current treatment options and novel nanotechnology-driven enzyme replacement strategies for lysosomal storage disorders. Advanced Drug Delivery Reviews, (2022); 114464.
- Yue WW, Mackinnon S, and Bezerra GA. Substrate reduction therapy for inborn errors of metabolism. Emerging Topics in Life Sciences, (2019); 3(1): 63-73.
- Höller A, Albrecht U, Sigl SB, Zöggeler T, Ramoser G, et al. Successful implementation of classical ketogenic dietary therapy in a patient with Niemann-Pick disease type C. Molecular genetics and metabolism reports, (2021); 27, 100723.
- Samaranch L, Pérez-Cañamás A, Soto-Huelin B, Sudhakar V, Jurado-Arjona J, et al. Ledesma MD. Adeno-associated viral vector serotype 9–based gene therapy for Niemann-Pick disease type A. Science Translational Medicine, (2019); 11(506): eaat3738.
- Massaro G, Geard AF, Liu W, Coombe-Tennant O, Waddington SN, Baruteau J, Gissen P, Rahim AA. Gene therapy for lysosomal storage disorders: ongoing studies and clinical development. Biomolecules, (2021); 11(4): 611.
- Jiang D, Lee H, and Pardridge WM. Plasmid DNA gene therapy of the Niemann-Pick C1 mouse with transferrin receptor-targeted Trojan horse liposomes. Scientific Reports, (2020); 10(1): 13334.
- Tran ML, Génisson Y, Ballereau S, and Dehoux CJM. Second-generation pharmacological chaperones: beyond inhibitors. Molecules, (2020); 25(14): 3145.
- Du J, Liu X, Zhang Y, Han X, Ma C, et al. The Effects of Combined Therapy With Metformin and Hydroxypropyl-β-Cyclodextrin in a Mouse Model of Niemann-Pick Disease Type C1. Frontiers in Pharmacology, (2022); 12: 825425.
- Holzmann C, Witt M, Rolfs A, Antipova V, and Wree A. Gender-specific effects of two treatment strategies in a mouse model of niemann-pick disease type c1. International Journal of Molecular Sciences, (2021); 22(5): 2539.
- Kurokawa Y, Osaka H, Kouga T, Jimbo E, Muramatsu K, et al. Gene therapy in a mouse model of niemann–pick disease type C1. Human Gene Therapy, (2021); 32(11-12): 589-598.
- Deodato F, Boenzi S, Taurisano R, Semeraro M, Sacchetti E, et al. The impact of biomarkers analysis in the diagnosis of Niemann-Pick C disease and acid sphingomyelinase deficiency. Clinica Chimica Acta, (2018); 486: 387-394.
- Jiang X, and Ory D. Advancing diagnosis and treatment of Niemann-Pick C disease through biomarker discovery. Exploration of Neuroprotective Therapy, (2021); 1(3): 146.
- Sobrido M-J, Bauer P, De Koning T, Klopstock T, Nadjar Y, Patterson MC, Synofzik M, Hendriksz CJ. Recommendations for patient screening in ultra-rare inherited metabolic diseases: what have we learned from Niemann-Pick disease type C? Orphanet Journal of Rare Diseases, (2019); 14(1): 1-14.
- Mengel E, Bembi B, Del Toro M, Deodato F, Gautschi M, et al. Clinical disease progression and biomarkers in Niemann-Pick disease type C: a prospective cohort study. Orphanet Journal of Rare Diseases, (2020); 15 (1): 1-18.
- Seaby EG, and Ennis S. Challenges in the diagnosis and discovery of rare genetic disorders using contemporary sequencing technologies. Briefings in Functional Genomics, (2020); 19(4): 243-258.
- Hammerschmidt TG, Ribas G. Saraiva-Pereira ML, Bonatto MP, Kessler RG, et al. Molecular and biochemical biomarkers for diagnosis and therapy monitorization of Niemann-Pick type C patients. International Journal of Developmental Neuroscience, (2018); 66: 18-23.
- Walterfang M, Di Biase MA, Cropley VL, Scott AM, O'Keefe G, Velakoulis D, Pathmaraj K, Ackermann U, Pantelis C. Imaging of neuroinflammation in adult Niemann-Pick type C disease: A cross-sectional study. Neurology, (2020); 94(16): e1716-e1725.
- Eskes EC, Sjouke B, Vaz FM, Goorden SM, Van Kuilenburg AB, et al. Biochemical and imaging parameters in acid sphingomyelinase deficiency: Potential utility as biomarkers. Molecular Genetics and Metabolism, (2020); 130(1): 16-26.
- Hammerschmidt TG, Encarnação M, Faverzani JL, de Fátima Lopes F, de Oliveira FP, et al. Molecular profile and peripheral markers of neurodegeneration in patients with Niemann-Pick type C: Decrease in Plasminogen Activator Inhibitor type 1 and Platelet-Derived Growth Factor type AA. Archives of Biochemistry and Biophysics, (2023); 735: 109510.
- Vujosevic S, Parra MM, Hartnett ME, O’Toole L, Nuzzi A, Limoli C, Villani E, Nucci P. Optical coherence tomography as retinal imaging biomarker of neuroinflammation/neurodegeneration in systemic disorders in adults and children. Eye (Lond), 2023. 37(2): 203-219.
- Bolukbasi S, Dogan C, Kiykim E, Cakir A, Erden B, et al. Multimodal imaging including optical coherence tomography angiography in patients with type B Niemann-Pick disease. International ophthalmology, (2019); 39: 2545-2552.
- Verheul C, Kleijn A, and Lamfers MLJ. Cerebrospinal fluid biomarkers of malignancies located in the central nervous system. Handbook of Clinical Neurology, (2018); 146: 139-169.
- Campbell K, Cawley NX, Luke R, Scott KE, Johnson N, et al. Identification of cerebral spinal fluid protein biomarkers in Niemann-Pick disease, type C1. Biomarker Research, (2023); 11(1): 14.
- Agrawal N, Farhat NY, Sinaii N, Do AD, Xiao C, et al. Neurofilament light chain in cerebrospinal fluid as a novel biomarker in evaluating both clinical severity and therapeutic response in Niemann-Pick disease type C1. Genetics in Medicine, (2023); 25(3): 100349.
- De Castro-Oros I, Irún P, Cebolla JJ, Rodriguez-Sureda V, Mallén M, et al. Assessment of plasma chitotriosidase activity, CCL18/PARC concentration and NP-C suspicion index in the diagnosis of Niemann-Pick disease type C: a prospective observational study. Journal of Translational Medicine, (2017); 15 (1): 43.
- Degtyareva AV, Proshlyakova TY, Gautier MS, Degtyarev DN, Kamenets EA, et al. Oxysterol/chitotriosidase based selective screening for Niemann-Pick type C in infantile cholestasis syndrome patients. BMC Medical Genetics, (2019); 20 (1): 123.
- Papandreou A, Doykov I, Spiewak J, Komarov N, Habermann S, et al. Niemann-Pick type C disease as proof‐of‐concept for intelligent biomarker panel selection in neurometabolic disorders. Developmental Medicine & Child Neurology, (2022); 64(12): 1539-1546.
- Maekawa M, Narita A, Jinnoh I, Iida T, Marquardt T, et al. Diagnostic performance evaluation of sulfate-conjugated cholesterol metabolites as urinary biomarkers of Niemann–Pick disease type C. Clinica Chimica Acta, (2019); 494: 58-63.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0