

Full Length Research Article
Association Mapping Study of Various Desirable Traits of Rice
Sumaira Aslam Chohan2, Muhammad Ashfaq1, Farah Khan*2
Adv. life sci., vol. 10, no. 2, pp. 249-258, June 2023
*- Corresponding Author: Farah Khan (drfarah_khann@yahoo.com)
Authors' Affiliations
2. Department of Botany, Lahore College for Women University, Lahore – Pakistan
[Date Received: 26/12/2022; Date Revised: 17/04/2023; Date Published: 30/06/2023]
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: This study was performed to evaluate the diversity of various morphological characters and their relationship with yield in rice. The goal of this work was to find quantitative trait loci (QTL) for yield, yield components, and other agronomic variables in 100 different rice germplasm samples, as well as to assess the genetic structure and degree of linkage disequilibrium in the rice germplasm diversity panel. To establish Linkage Disequilibrium (LD) between markers and causative mutations, marker density is essential. Linkage disequilibrium (LD) patterns of various SNP markers on all chromosomes. If markers are sufficiently dense to have good coverage of LD, the LD decay with distance can be compared to the marker density.
Methods: Different traits were measured and recorded under Randomized Complete Block Design (RCBD) experiment. DNA extraction and PCR analysis was done to measure the genotypic characteristics of rice. Genotypic and phenotypic variability was measured by using ANOVA and GWAS.
Results: For pair-wise markers, linkage disequilibrium is calculated as R square and plotted versus the distance between the markers. In this study, the overall phenotypic variability among the examined traits was represented by R2 and ranged from 11.47% to 25.44%. The genetic architecture of these traits may be implied by the recently identified genomic regions (loci). An influential replacement for bi-parental gene maps, genome-wide association studies (GWAS) use data from genome-wide markers in large amounts of easily obtained germplasm.
Conclusion: The linkage disequilibrium, which is the non-random link between an allele at two or more loci, is used in this mapping method to infer the innate relationships between phenotypic variations and marker polymorphisms. Genome Wide Association Study (GWAS) of genotypes provides the information about for the selection of genotypes and determination of new marker trait association.
Keywords: Oryza sativa L; Rice; DNA; Association mapping; Traits
Introduction![]()
Rice (Oryza sativa L) belongs to the Poaceae family also known as “Gramineae”, and subfamily Orizoideae, and is an essential food for about one-third population around globe, occupying, about one fifth of arable land covered by cereals [1]. It has been grown in many geographical areas under a variety of cultural conditions. Maximum of the rice in world is produced and consumed in Asia, accounting for approximately more than fifty percent of the world’s population. About 11% of the total arable land around the globe is seeded with rice annually, second only to wheat, providing twenty percent of the everyday calories to the people on earth [2]. However, to feed the growing world’s population with inadequate arable land it is necessary to attain high rice yielding varieties by improving flowering time, and yield related traits [3]. However, over the past 25 years, the output of rice worldwide has doubled, partly as a result of the application of enhanced technologies, including maximum grain producing varieties and enhanced crop development techniques [4].
Efficient tillers, quantity of seeds per panicle with thousand grain weight (TGW) are the key factors in measuring yield of rice. The grain length (GL) [5, 6] and grain width (GW), [7,8,9,10,11], grain filling [12], flowering (heading date, HD) are also important for production of high-yielding rice cultivars [13].
The production of rice must be expanded to ensure food security globally. To tackle the challenge posed by a constantly growing population, crossbreeding technology has a significant role. Hybrid rice outperforms the best traditional varieties in large-scale cultivation around the world by 15-20% (or almost more than 1 ton of rice/hectare) [14]. Production of hybrid rice seeds at commercial level is essential for the successful adoption of hybrid rice, because it is a self-pollinated crop [15]. Potential for crop development will increase with the use of contemporary biotechnological techniques and the preservation of diversity at genetic level and variability in plant breeding practices. The potential for future crop development is greatly increased by the high genetic diversity found both within and between rice cultivars and their wild relatives.
Rice has a large amount of genetically diverse material that is well-conserved (about 100,000 accessions of germplasm around the globe), a small genome size of 430 Mb [16, 17], diploid genetics and a high level of genomic polymorphism [1, 18, 19].
Traditional selection-based breeding is challenging due to the tiny mass of spikelets and the major environmental influence on blooming in rice. As a result, there has been an increase in interest in creating DNA markers linked to desirable qualities for breeding programmes [20]. Therefore, estimating and measuring the genetic variation present in the rice germplasm is necessary for improving its genetic makeup.
The use of phenotypic and biochemical markers allowed the assessment of genomic diversity and the creation of a connection between cultivars. They are less popular among researchers since they are stage-specific, minor in number, and greatly affected by the environment. A new era in crop plant genetic characterization has begun with the development of PCR reliant genetic marker technologies. Because of their great simplicity and repeatability, straightforward ability to score, dependability, co-dominant character, and multiple allelic makeup. Microsatellite (SSR) markers are a popular technique among many molecular markers. Microsatellites, which are arrangements of a small number of repeated and close by DNA base pairs, are widely distributed in the genome of eukaryotes [21]. By creating primers (20–30 base pairs) corresponding to conserved sequences surrounding the microsatellite and specifically tailored for amplification, polymerase chain reaction (PCR) can be utilized to identify differences in the number of repeats. Numerous genetic studies on rice have used these markers for genotypic analysis, molecular phylogenetic evaluation and comparative genomic research [22, 23]. According to a notion, the functionally favorable alterations at the coding sections of the genome, which are necessary for breeding programmes, would not be adequately captured by the usage of randomly selected markers to quantify genetic variation [6]. The objective of the study was to determine the phenotypic and genotypic variability on the basis of yield and yield related traits.
Methods![]()
Screening of Germplasm
In the experimental area of the Institute of Agricultural Sciences, University of the Punjab, Lahore, 100 rice germplasm lines/varieties were gathered from the rice research institute Kala Shah Kaku in Pakistan, NARC in Islamabad in Pakistan, and USDA in Arkansas in the United States. Forty different parents were chosen based on plant and seed morphological characteristics that were verified using a variety of Floras [24-26].
Phenotypic Characters
The following plant characteristics are listed for each plant species: plant height (cm), productive tillers/plant, flag leaf area (m2), panicle length (cm), days to 50% heading, days to maturity, yield per plant (g), and harvest index (%). The dimensions of a seed include its width (mm), length (mm), length to width ratio, thickness (mm), weight (g) per panicle (1000 seeds), and the proportion of fertile seeds in each panicle etc.
Genotyping Rice Lines Using Microsatellite Markers
A total of 100 rice accessions were raised in experimental area of the Institute of Agricultural Sciences, University of the Punjab, Lahore. Young leaves from the accessions were collected for genomic DNA extraction.
Isolation of genomic DNA
Rice leaf samples were collected, and genomic DNA of each rice line was extracted by following the CTAB method illustrated by Doyle and Doyle, 1987 [27].
PCR amplification
The reaction mixture (15 μl) for PCR amplification was prepared by adding DNA 25 ng/μl (0.60μl), dNTPs (2.5 mM) (2μl), forward and reverse primers (1.5μl each), Taq polymerase (0.10μl of 3units/μl) and sterile water (7.80μl) to make final concentration of 15 μ. The reaction mixture was mixed by a vortex for 30 seconds to mix the components. Then PCR tubes (200 µl) were loaded in a thermal cycler. The reaction in thermal cycler (Eppendorf Master Cycler Gradient) program consisted of 6 cycles, which included Initial denaturation at 94˚C for 5 minutes, Denaturation at 94˚C for 1 minute, annealing at 55˚C for 1 minute, Extension at 72˚C for 1 minute (36 repeated cycles of denaturation, annealing and extension) and Final extension at 72˚C for 5 minutes and holding the samples for 5 minutes at 4˚C.
Agarose gel electrophoresis
Agarose gel (1.2%) electrophoresis was performed to separate PCR amplified products by dissolving 1.2 g agarose in TE buffer by heating for 2 minutes in microwave. The gel was allowed to cool for a minute and 3 μl of ethidium bromide was added to it. The gel was poured on gel casting tray supplied with comb. The DNA sample with 6x loading dye were loaded, 1 kb DNA ladder and one blank sample were also loaded as control. Gel was allowed to run at 100V for 50 minutes. The gel was observed on gel documentation system under UV light.
The 100 rice accessions were genotyped using a total of 100 SSR primers, as previously mentioned. To investigate the marker-trait linkage, phenotypic data from field trials and genome-wide polymorphic SSR marker data were combined. Excel's base format was used to complete the analysis of variance (ANOVA). Using the programme STRUCTURE version 2.2, the population's genetic structure (Q) was predicted and clustered [28]. TASSEL v2.0.1 was used for the association analysis, with 50000 mutations per mutation used to correct for multiple testing [28]. Markers were considered significant if the adjusted P value was greater than 0.05.
Results![]()
Association Mapping using SSR
For all characteristics in the two growth seasons (2018-19 and 2019-20) there was considerable phenotypic and genotypic variance among the 100 rice genotypes. Polymorphic SSR markers were employed in the study to genotype the 100 rice accessions. To find the significant markers for the 18 investigated characteristics, 216 polymorphic SSR markers were employed in the association mapping method. TASSEL software was used to find these microsatellite markers that were associated with the investigated phenotypic attributes. This study identified a total of 201 marker-trait associations (MTAs) at P0.0001 significant levels after crossing the false discovery rate (FDR) at less than 0.05.
Plant Height
In association analysis, 16 SSR markers were detected to be considerably associated with plant height, which are located on chromosomes 4, 6, 8, 9, 10, 11 and 12. These markers, namely RM5642, RM1321, RM1986, RM5755, RM2884, RM1113, RM5988, RM5708, RM6487, RM2935, RM1261, RM6475, RM6623, RM3153, RM5752, and RM3137, were appreciably related with this trait. A range of 11.15% to 28.57% of the total phenotypic variance (PV) was explained by the markers linked with plant height. In this study, the SSR marker (RM3137) from chromosome 11 (position 32.7cM) indicated the lowest value (11.15%) of trait variability and the marker (RM5642) from chromosome 5 (position 91.2cM) represented the maximum value of trait variability (28.57%).
Productive Tillers/Plant
Total thirteen SSR markers were found to be strongly linked with Productive Tillers/Plant. Out of these, two markers were from chromosomes 4, 6 and 8 and the remaining were from chromosomes 1, 2, 3, 5, 9, 11 and 9. Total PV for this trait using these markers varied from 10.59% to 20.67%. In this experiment, the marker (RM5665) from chromosome 3 (position 158.2cM) exhibited the highest PVE (20.67%), whereas the marker (RM5638) form chromosome 1 (position 86cM) gave the lowest PVE (10.59%).
Flag Leaf Area
Flag leaf area was highly associated with eleven SSR markers. The SSR markers namely RM5786, RM5642, RM1002, RM5988, RM5654, RM5686, RM3437, RM6314, RM4710 RM5900, RM5666 and RM3153 showed significant association with this trait. The overall phenotypic variation in this characteristic was explained by these markers by 11.48% to 23.18%. On chromosome 9 (position 77.2cM), the marker (RM5786) described the greatest amount of phenotypic trait variability (23.18%), whereas the marker (RM5666) on chromosome 10 (position 72.5cM) explained the least amount (11.48%). In this work, 9 chromosomes were used as markers for trait association for flag leaf area. These markers included 3 SSRs at chromosome 5, 2 SSRs at each of chromosomes 3 and 4, and 1 SSR at each of chromosomes 2, 6, 9 and 10.
Panicle Length
Total ten important SSR markers, including three on chromosome 5, two on chromosome 12, and the rest on chromosomes 2, 8, 9, 10, and 11, were found to be substantially associated with the panicle length. From 10.18% to 24.69% of the phenotypic variation for panicle length was explained by these markers. While the marker (RM5731) on chromosome 11 (place 54.3cM) explained the least variation (10.18%) in the panicle length, the marker (RM5651) on chromosome 2 (position 93.1cM) explained the most variation (24.69%). The significant associated markers with panicle length were RM5651, RM4837, RM5642, RM3351, RM1986, RM1235, RM1026, RM5887, RM3326 and RM5731.
Spikelets per Panicle
Eleven SSR markers, two of which were found on each of chromosomes 3, 5, 10, and 11, and the other three on chromosomes 1, 8, and 9, were substantially related with the panicle length. The significantly associated markers were RM4837, RM5686, RM5756, RM5704, RM2326, RM5374, RM8019, RM5666, RM1812, RM1195 and RM5657 with spikelets per panicle. The phenotypic variance in the spikelets per panicle was explained by all of these SSRs in the range of 14.53% to 29.78%. On chromosome 5 (position 40.6cM), the marker (RM4837) described the most variation (29.78%) and on chromosome 9 (position 49.3cM), the marker (RM5657) explained the least variation (14.53%) in the spikelets per panicle.
Number of Primary Branches per Panicle
The number of primary branches per panicle was found to be correlated significantly with sixteen SSR markers. Eight chromosomes contained MTA for NG, including 4 SSRs on chromosome 2 and 2 SSRs on chromosomes 3, 4, 9, and 12, whereas the remaining chromosomes included MTA for chromosomes 1, 5, 8, and 10. The markers RM5786, RM5651, RM5665, RM5654, RM6314, RM1321, RM1328, RM3351, RM1235, RM1063, RM1319, RM5709, RM2935, RM1986, RM5631 and RM5708 showed significant association with various morphological traits of rice. The phenotypic variance in the branches per panicle was explained by all of these SSRs in the range of 11.93% to 26.85%. For the number of primary branches per panicle in 100 rice genotypes, the marker (RM5708) on chromosome 10 (position 30.2cM) explained the least variation (11.93%) and the marker (RM5786) on chromosome 9 (position 77.2cM) explained the most variation (26.85%).
Number of Secondary Branches per Panicle
In association analysis, 13 SSR markers were observed to be considerably associated with number of secondary branches per panicles. Three SSR markers were at chromosome 12, 2 SSRs on each of the chromosomes 2, 6 and 8, while other SSRs on the chromosomes 3, 5 9, 10. The markers RM8270, RM7217, RM5755, RM1113, RM3262, RM3295, RM8107, RM3326, RM5900, RM5353, RM1261, RM5657 and RM6288 were significantly associated with number of secondary branches per panicles. Phenotypic variation (PV) in the number of secondary branches per panicle associated markers accounted for 13.01% to 26.97% of the total phenotypic variation (PV) for this trait. Marker (RM6288) from chromosome 12 (position 13.3cM) described the minimum variability (13.01%) of secondary branches per panicle, while the marker RM8270 from chromosome 6 (position 35.8cM) indicated the maximum value of this trait’s variability (26.97%).
Days to 50% Heading
Total 12 SSR markers (RM3688 followed by RM5651, RM5665, RM5642, RM4837, RM3351, RM3330, RM1328, RM1986, RM6881, RM4710 and RM5666) were strongly associated with days to 50% heading. Total PV for this trait using these markers ranged from 12.76% to 22.93%. The marker RM3688 on chromosome 6 (position 88.2cM) exhibited the highest PVE (22.93%), whereas the marker RM5666 on chromosome 10 (position 72.5cM) explained the least percentage (12.76%) of the days to 50% heading in the analyzed germplasm.
Days to Maturity
Days to maturity was highly associated with nine SSR markers (RM3351, RM5642, RM5988, RM1002, RM3437, RM4710, RM8270, RM3133 and RM5756) and the overall PV ranged from 12.44% to 20.16%. On chromosome 5 (position 80.7cM), the marker RM3351 explained the most phenotypic trait variability (20.16%), while on chromosome 10 (position 48.4cM), the marker RM5756 explained the least variability (12.44%). Nine chromosomes were involved in the distribution of markers trait association for days to maturity, including 4 SSRs at chromosome 5, 2 SSRs at chromosome 6, and the remaining SSRs at chromosomes 3, 10, and 11.
Yield per Plant
There were 10 significant SSR markers in all, and two of them were found on each of the chromosomes 5 and 6, while the others were found on chromosomes 2, 3, 4, 9, 11, and 12. These markers accounted for 11.83% to 17.36% of the phenotypic variation in yield per plant. While the marker RM5367 on chromosome 11 (position 0.0cM) explained the least variation (11.83%) for yield per plant, the marker RM4837 on chromosome 5 (position 40.6cM) explained the most variation (17.36%). The significant associated markers with this trait were RM4837, RM5642, RM3330, RM5665, RM5786, RM1986, RM5988, RM5709, RM5631 and RM5367.
Harvest Index
Ten important SSR markers (RM1986, RM5686, RM5654, RM1321, RM4837, RM1002, RM6314, RM7217, RM5755 and RM5756) that were significantly associated with the harvest index were identified. The PV in them ranged from 15.07% to 21.98%. MTA for the harvest index were spread out throughout 10 chromosomes, with 3 SSRs on chromosome 3 and 2 SSRs on each of chromosomes 10 and 11 and the others on chromosomes 1, 2, 4, 5, and 12. For the harvest index of 100 rice genotypes, the marker RM1986 on chromosome 12 (position 73cM) described the most phenotypic variation (21.98%), whereas the marker (RM5756) on chromosome 10 (position 48.4cM) explained the least variation (15.07%).
Number of Fertile Seeds per Panicle
The quantity of fertile seeds per panicle was substantially correlated with the presence of fourteen significant SSR markers; four on chromosome 5, three on each of chromosomes 1, 2, and 10, two on each of chromosomes 1, 2, and 9, and one on chromosome 4. These markers accounted for 12.53% to 27.34% of the phenotypic variation in the quantity of viable seeds per panicle. The largest variance (27.34%) was explained by the marker (RM1054) on chromosome 5 (position 40.6cM), while the minimal variation (12.53%) was explained by the marker (RM1054) on chromosome 5 (position 122cM). The significant associated marker with number of fertile seeds per panicle were RM4837, RM5708, RM4710, RM5374, RM3337, RM3340, RM5666, RM1068, RM6824, RM5661, RM1038, RM2482, RM1095 and RM1054
Seeds Weight per Panicle
Nine SSR markers were highly correlated with seeds weight per panicle, with two SNPs on each of chromosomes 1 and 4 and the other four on chromosomes 2, 3, 5, 9, and 10. The significantly associated markers were RM5686, RM5642, RM1328, RM6487, RM1113, RM1032, RM1038, RM5887 and RM1117 with seeds weight per panicle. The phenotypic variance in this characteristic was explained by all of these SSRs in the range of 11.57% to 19.74%. While the marker RM1117 on chromosome 1 (position 122.6cM) described the least variation (11.57%) in seeds weight per panicle, the marker RM5686 explained the most variation (19.74%) on chromosome 3 (position 40.3cM).
Seed Width
Six important SSR markers were linked with seed width in this investigation. Six chromosomes were used to distribute MTA for seed width, with three SSRs on chromosome 5 and the remaining ones on chromosomes 2, 6, and 9. The markers RM4837, RM5642, RM5988, RM4710, RM1092 and RM5786 showed significant association with seed width. The phenotypic variance in this characteristic was explained by all of these SSRs in the range of 10.0% to 14.71%. In 100 rice genotypes, the marker RM5786 on chromosome 9 (position 77.2cM) described the least variation (10.0%) for seed width, whereas the marker RM4837 on chromosome 5 (position 40.6cM) explained the most phenotypic variation (14.71%).
Seed Length
Eight SSR markers (RM5651, RM5665, RM1063, RM6824, RM5988, RM5708, RM2859 and RM1032) were observed to be strongly linked with seed length using association analysis. Eight chromosomes in total included 8 important SSRs, with 2 SSRs on each of chromosomes 2 and 10 and the remaining SSRs on 1, 3, 6, and 11. The percentage of the total phenotypic variation (PV) seed length ranged from 11.06% to 19.53%. The marker RM5651 from chromosome 2 (position 93.1cM) indicated the maximum value of trait variability (19.53%), whereas the marker RM1032 from chromosome 1 (position 49.3cM) described the minimum value (11.06%) of seed length variability.
Seed Length to Width Ratio
Total 10 SSR markers (RM5686, RM5709, RM1125, RM8270, RM6862, RM5887, RM5708, RM5816, RM1038 and RM2859) were strongly associated with Seed Length to Width Ratio. Total PV for this trait by these markers varied from 12.17% to 24.22%. In the examined germplasm, the marker RM5686 on chromosome 3 (position 40.3cM) had the highest PVE (24.22%), whereas the marker RM2859 on chromosome 11 (position 89cM) accounted the least amount (12.17%) of the Seed Length to Width Ratio.
Seed Thickness
Seed thickness was highly associated with twelve SSR markers (RM6487, RM5756, RM6881, RM5631, RM3337, RM1817, RM5353, RM1068, RM5847, RM3320, RM2705 and RM662). The entire phenotypic variance for this characteristic was explained by these seed thickness-related markers in the range of 12.73% to 28.68%. On chromosome 4 (location 12.2cM), the marker RM6487 explained the most phenotypic trait variability (28.68%), while on chromosome 12 (position 101.9cM), marker RM6623 explained the least PV (12.73%). Seed thickness trait markers were found to be dispersed throughout 8 chromosomes, with 3 SSRs at each chromosome 9 and 2 SSRs at chromosome 4, and the remaining SSRs at chromosomes 1, 2, 3, 7, 8, and 10.
1000 Seed Weight
Total eleven significant SSR markers (RM1986, RM1328, RM5756, RM1125, RM8107, RM5531, RM5704, RM3083, RM1106, RM8219 and RM3326) were strongly linked with 1000-seed weight including, 2 markers located on each chromosome 6, 10, 11, 12 and the others on 2, 4 and 9 chromosomes. These markers explained 19.74 % to 29.88 % of the phenotypic variation for 1000-seed weight. The marker RM1986 on chromosome 12 (position 73cM) explained maximum variation 29.88 %, while the marker RM3326 on the same chromosome explained minimum variations (19.74%) for 1000-seed weight.
Figures & Tables
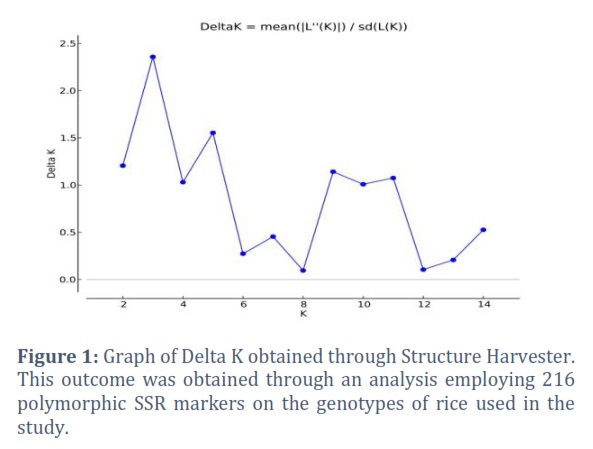
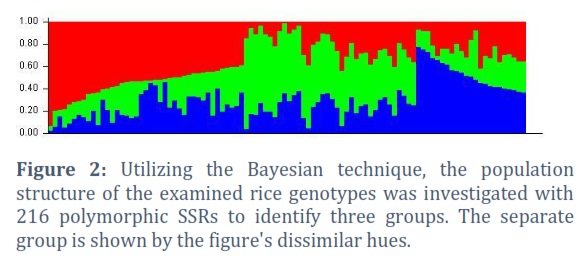
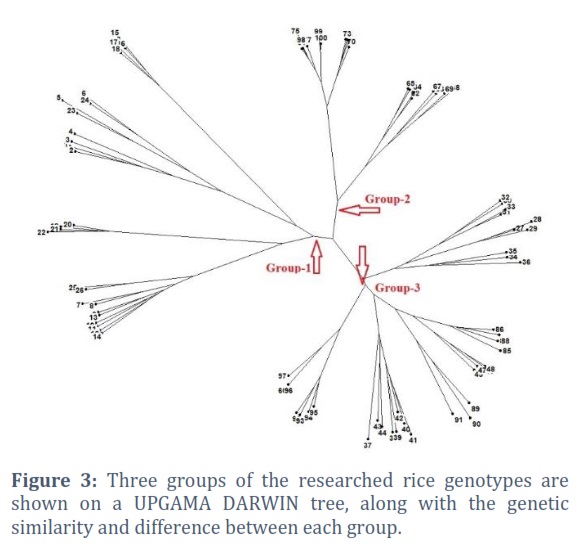
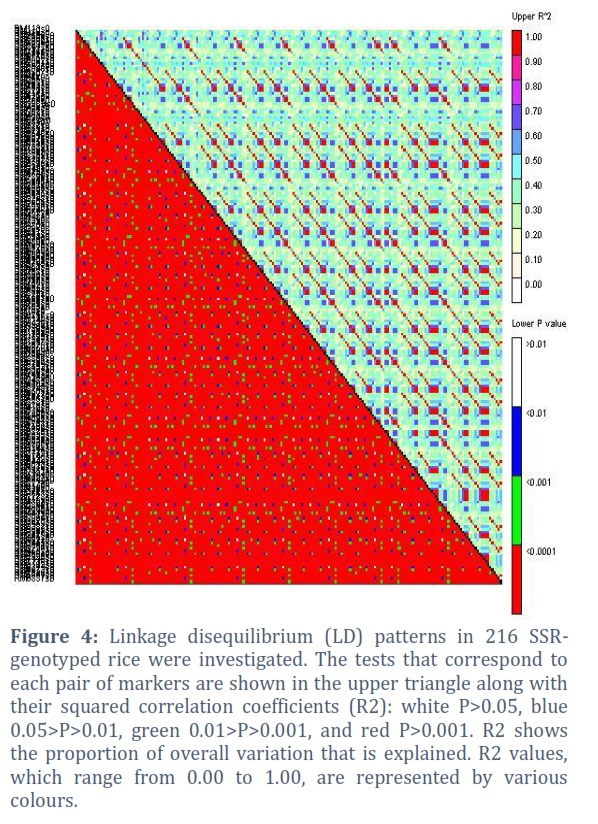
To determine the quantitative trait loci (QTL) for yield, yield components, and other agronomic variables in 100 different rice germplasm samples, as well as to assess the genetic structure and degree of linkage disequilibrium in the rice germplasm diversity panel. During the two years 2018–19 and 2019–20, phenotypic evaluation of yield and agronomic traits was carried out for this aim. For GWAS analysis utilising a 10k SNP array, average values for each trait were calculated using data from both years and used by running GAPIT in the R software. To establish Linkage Disequilibrium (LD) between markers and causative mutations, marker density is essential (Figure 4).
During plant height analysis, total 31 SNPs were found to be significantly associated with this trait which were located on chromosomes 1, 2, 4, 6, 8, 9, 10, 11 and 12 . Phenotypic variation explained (PVE) by plant height associated markers exhibited from 10.61 % to 17.58% of the total phenotypic variation. In GWAS using 100 rice genotypes, total eight SNPs were strongly associated with productive tillers/plant. Out of these, there were one SNP at chromosome 10, two at chromosome 3, one at each chromosome 6, 7, and 11, whereas 2 MTAs were at chromosome 12. The overall phenotypic variance for plant height explained by these markers ranged from 16.44% to 19.42%. 17 SNP markers had a strong correlation with flag leaf area. These MTAs accounted for 13.83% to 28.59% of the phenotypic variance overall in this characteristic. On chromosome 4 at 24529999 cM, the SNP marker OsGRg06715 described the most phenotypic trait variability (28.59%), whereas on chromosome 3 at 34431137 cM, the marker OsGRg06026 explained the least variation (13.83%). Only three significant MTAs were observed for panicle length, with 1 SNP located at each chromosome 2, 8 and 3. These SNPs explained 11.28% to 14.96% of the phenotypic variation for panicle. Two important MTAs were discovered to be connected to each spikelet per panicle. Between 25.75% and 29.88% of the overall phenotypic variability was explained by these SNPs. Only 2 MTAs were discovered to be substantially linked with the number of primary branches per panicle by association analysis. These important SNPs were split between two chromosomes, one on each of the chromosomes 6 and 10. Total phenotypic variation explained by the number of primary branches per panicle associated with SNPs was from 19.83% to 28.70% [29-33].
Total three MTAs were strongly associated with number of secondary branches per panicle. Out of these 1 SNP was from each of the chromosomes 6 (OsGRb26900), 8 (OsGRb28010) and 9 (OsGRg12400). Total PV in the number of secondary branches per panicle by these SNPs ranged from 17.98 % to 25.75 %. Days to 50% heading was highly associated with twelve SNP markers. The SNPs showed significant association with this trait, which were located on chromosomes numbers 7, 4, 2, 6, 9 12 and 11. The Days to 50% heading related markers explained from 12.31 % to 24.14 % of the total phenotypic variation in the number of secondary branches per panicle. Total twelve significant MTAs were strongly linked with days to maturity. Overall these significant SNPs were distributed across 8 chromosomes; out of which, 1 MTA was on each of the chromosomes 3, 5, 9, 11 and 12, 2 SNPs on each of the chromosomes 2 and 7, while three SNPs on chromosomes 6. These SNP markers explained from 23.51 % to 30.72% of the phenotypic variation for days to maturity. Ten significant MTAs were observed for the yield per plant, including, 1 SNP located on each of the chromosomes 6 and 1, 4, one significant SNP on chromosome 3, while 2 SNPs on each of the chromosomes 7 and 11. These SNPs explained 18.97% to 29.55% of the phenotypic variation for yield per plant.
Harvest index that accounted for 19.07% to 26.06% of the total phenotypic variation was highly associated with only 2 SNP markers. The SNP marker OsGRb19717 explained maximum phenotypic trait variability (26.06%) on chromosome 10 at 117305836cM and the marker OsGRb24377 on chromosome 1 at 40794243cM explained minimum variability (19.07). In association analysis, 7 MTAs were found to be significantly associated with number of fertile seeds per panicle. All these significant SNPs were distributed among 4 chromosomes. 1 SNP on each of the chromosomes 8 and 7, 2 on chromosome 6 and 3 on chromosome 5. Phenotypic variation, explained by the number of fertile seeds per panicle, varied from 20.96% to 30.72%. In total, 13 significant MTAs were noticed for seed weight per panicle, having 4 SNPs located on each of the chromosomes 2 and 1 and 2 significant SNPs on each of the chromosomes 10 and 3, whereas chromosome 6 had only 1 MTP. These SNPs explained 14.96% to 26.64% of the phenotypic variation for seeds weight per panicle
In GWAS, using 100 rice genotypes, total nine SNPs were strongly associated with seed width. Out of them, five SNPs were from chromosomes 12, one from each of the chromosomes 2, 11, 3, 6. Total phenotypic variation explained by these markers ranged from 11.72 % to 22.61 % for seed width. Ten MTAs were strongly associated with the seed length, out of which, two SNPs were from each of the chromosomes 11, 4 and 9, 1 from chromosome 10 and 3 from chromosome 3. Total PV for seed length by these SNPs ranged from 14.96 % to 22.05 %. A total of 11 SNPs were found to be significantly associated with ratio of seed length to its width which were located on chromosomes 10, 4, 11, 1, 3 and 6. Phenotypic variation explained (PVE) by the seed length to width ratio with associated markers exhibited from 13.70 % to 26.14% of the total phenotypic variation [34-38]
In association analysis, only 2 MTAs were found to be significantly associated with seed thickness, which were distributed across two chromosomes, 1 on each of the chromosomes 6 and 1. Eleven significant SSR markers were strongly linked with 1000-seed weight including 2 markers located on each of the chromosomes 6, 10, 11, 12 and the others on 2, 4 and 9 chromosomes These markers explained 19.74% to 29.88 % of the phenotypic variation for 1000-seed weight. Similar study was reported by other scientists [39-45].
The genetic source of the rice breeding material investigated in this study was estimated using the Bayesian approach, and the findings revealed the peak value of K=3, suggesting the presence of three subgroups among the tested rice germplasm (Figure 1). With the use of 204 polymorphic SSR markers from a Structure Harvester analysis, this result was obtained by using 100 rice genotypes. It is based on the maximum likelihood estimation of your model's variance given a certain K and a second order derivation of that variance. Only the highest level of clustering and the total number of subpopulations in the main population are shown by delta K. STRUCTURE Software exhibited a coloured graph to show the three subpopulations, with each group being represented by a distinct colour (Figure 2). Along with other statistical techniques, such as population structures, clustering many germplasms is a quick and efficient method for determining the genetic diversity of germplasm [28, 46] and DARWIN analysis [47]. Using STRUCTURE analysis, 100 rice genotypes were divided into 3 sub-populations in our study as indicated in Figure 3. This is the indication that, based on the molecular data, the rice accessions were considerably divided into three sub-populations. Population structure results from deviations from random mating in the sampling population causes certain people to be more linked to one another than the others. The assumption of independent mistakes in the assessment of allele effects using ordinary least squares is incompatible with population structure [22].
Suvi [48] provided the population structure of many rice types from diverse origins in a prior study. Courtois [49] identified two subgroups and classified rice types into two groups with little mixing lines. A total of 416 rice genotypes from China had seven subpopulations according to Ali [50]. A collection of 91 accessions of rice germplasm from eastern and northeastern India have been divided into four groups by population structure analysis by Suvi [48]. This study's structural analysis revealed that the investigated germplasm was separated into three groups and that there was evidence of genetic variety within studied germplasm, clearly demonstrating that these genotypes have various genetic origins. The 100 rice genotypes were also divided into three subgroups via the DARWIN tree (UPGMA cluster) analysis.
The study would be very useful for the selection of genotypes on the basis of variability among the traits. Genome Wide Association Study (GWAS) of genotypes provides the information about for the selection of genotypes and determination of new marker trait association. Different MTAs were screened with respect to their respective traits. Number of genotypes have been identified based on various yield and related traits. Such types of association would be very useful in future breeding programs for the improvement and development of new plant populations with high genetic potential in the changing climatic conditions. For indirect selection, quantitative traits such as seeds per panicle, seed weight per panicle, panicle length, 1000 grain weight, yield per plant and yield per hectare could be used for the screening of high yielding rice genotypes.
Conflict of Interest
The authors declare that there is no conflict of interest.
Acknowledgement
The authors are very thankful to Lahore College for Women University (LCWU) and University of the Punjab providing funds for the completion of this study.
![]()
References
- Chakravarthi BK, Naravaneni R. SSR marker based DNA fingerprinting and diversity study in rice (Oryza sativa. L). African Journal of Biotechnology, (2006); 5(9): 684-688.
- FAO, (Food and Agriculture Organization of the United Nations) (2004) Rice is life. International Year of Rice, Rome, Italy
- Xue W, Xing Y, Weng X, Zhao Y, Tang W, Wang L, Zhou H, Yu S, Xu C, Li X. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nature Genetics, (2008); 40: 761–767.
- Byerlee, D. 1996. Knowledge-Intensive Crop Management Technologies: Concepts, Impacts, and Prospects in Asian Agriculture. International Rice Research Conference, Bangkok, Thailand, 3-5 June, 1996.
- Fan C, Xing Y, Mao H, Lu T, Han B, Xu C, Li X, Zhang Q. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theoretical and Applied Genetics, (2006); 112: 1164–1171.
- Zhang P, Liu X, Tong H, Lu Y, Li J. Association mapping for important agronomic traits in core collection of rice (Oryza sativa L.) with SSR markers. PLOS ONE, (2014); 9: e0111508.
- Song XJ, Huang W, Shi M, Zhu MZ, Lin HX. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nature Genetics, (2007); 39: 623–630.
- Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, Konishi S, Yano M. Deletion in a gene associated with grain size increased yields during rice domestication. Nature Genetics, (2008); 40: 1023–1028.
- Li XY, Qian Q, Fu ZM, Wang YH, Xiong GS, Zeng DL, Wang XQ, Liu XF, Teng S, Hiroshi F, Yuan M, Luo D, Han B, Li JY. Control of tillering in rice. Nature, (2003); 422: 618–621.
- Wang E, Wang J, Zhu X, Hao W, Wang L, Li Q, Zhang L, He W, Lu B, Lin H. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nature Genetics, (2008); 40: 1370–1374.
- Sun LJ, Li XJ, Fu YC, Zhu ZF, Tan LB, Liu FX, Sun XY, Sun XW, Sun CQ. GS6, a member of the GRAS gene family, negatively regulates grain size in rice. Journal of Integrative Plant Biology, (2013); 55: 938–949.
- She KC, Kusano H, Koizumi K, Yamakawa H, Hakata M, Imamura T, Fukuda M, Naito N, Tsurumaki Y, Yaeshima M, Tsuge T, Matsumoto K, Kudoh M, Itoh E, Kikuchi S, Kishimoto N, Yazaki J, Ando T, Yano M, Aoyama T. A novel factor FLOURY ENDOSPERM2 is involved in regulation of rice grain size and starch quality. The Plant Cell, (2010); 22: 3280–3294.
- Fei-fei UX, Liang J, Yan H, Chuan T, Ya-ling C, BAO Jin-song AOB. Association mapping of quantitative trait loci for yield-related agronomic traits in rice (Oryza sativa L.) Journal of Integrative Agriculture, (2016); (10): 2192-2202.
- Xu Y, Beachel H, McCouch SR. A Marker_ Based Approach to Broadening the Genetic Base of Rice in the USA. Crop Sciences, (2004); 44: 1947–1959.
- Virmani, S.S. Heterosis and hybrid rice breeding. Monographs Theoretical and Applied Genetics. 22. 1994, Springer-Verlag.
- Causse MA, Fulton TM, Cho YG, Ahn SN, Chunwongse J, Wu K, Xiao J, Yu Z, Ronald PC, Harrington SE, Second G, McCouch SR, Tanksley SD. Saturated molecular map of the rice genome based on a interspecific backcross population. Genetics, (1994); 138: 1251- 1274.
- Kurata N, Nagamura Y, Yamamot K, Harushima Y, Sue N, Wu J, Antonio BA, Shomura A, Shimizu T, Lin SY. A 300 kilobase interval genetic map of rice including 883 expressed sequences. Nature Genetics, (1994); 8: 365-372.
- Mccouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, Declerck G, Schneider D, Cartinhour S, Ware D, Stein L. Development Of 2240 New Ssr Markers For Rice (OryzaSativa L.). DNA Research, (1994); 9(6): 199-207.
- Tanksley S.D. Molecular markers in plant breeding. Plant Molecular Biology Reproduction, (1989); 1: 3-8.
- Yan GW, Li Y, Agrama AH, Luo D, Gao F, Lu X, Ren G. Association mapping of stigma and spikelet characteristics in rice (Oryza sativa L.). Molecular Breeding, (2009); 24: 277–292.
- Powell W, Machray GC, Provan J. Polymorphism revealed by simple sequence repeats. Trends in Plant Sciences, (1996); 1: 215–222.
- Babu BK, Meena V, Agarwal V, Agrawal P. Population structure and genetic diversity analysis of Indian and exotic rice (Oryza. sativa L.) accessions using SSR markers. Molecular Biology Reports, (2014); 41:4329-4339.
- Varaprasad GS Rani NS. Assessment of genetic diversity among basmati and non-basmati aromatic rices of India using SSR markers. Current Science, (2010); 99(2): 221–226.
- Hooker, J.D. The Flora of British India. L. Reeve & Co. publisher, 1885, London.
- Tutin T, Heywood G. Flora of Europe. Cambridge University Press, 1972.
- Nasir, E.; Ali, S.I.. Flora of Pakistan, Poaceae. Thomas, A. Cope. Herbarium Royal Botanical Garden Kew, England, 1982, p. 680.
- Doyle JJ, Doyle JL. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochemical Bulletin, (1987); 19: 11-15.
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics, (2000); 155(2): 945-959. doi: 10.1093/genetics/155.2.945. PMID: 10835412; PMCID: PMC1461096.
- Zhou JA, You Z, Ma L, Zhu G, He. Association analysis of important agronomic traits in japonica rice germplasm. African Journal of Biotechnology, (2012); 11: 2957–2970.
- Agrama AH, Eizenga CG, Yan W. Association mapping of yield and its components in rice cultivars. Molecular Breeding, (2007); 19: 341–356
- Wen WH, Mei F, Feng S, Yu, Huang J, Wu L, Chen X, Xu L, Luo. Population structure and association mapping on chromosome 7 using a diverse panel of Chinese germplasm of rice (Oryza. sativa L.). Theoretical and Applied Genetics, (2009); 119: 459–470.
- de Oliveira BT, Brondani R, Breseghello F, Coelho A, Mendonca J, Rangel P, Brondani C. Association mapping for yield and grain quality traits in rice (Oryza. sativa L.). Genetics and Molecular Biology, (2010); 33: 515-524.
- Ashfaq M, Rasheed A, Sajjad M, Ali M, Rasool B, Javed MA, Allah SU, Shaheen S, Anwar A, Ahmad MS, Mubashar U. Genome wide association mapping of yield and various desirable agronomic traits in Rice. Molecular Biology Reports, (2022); 49(12):11371-11383.
- Horii H, Nemoto K, Miyamoto N, Harada J. Quantitative trait loci for adventitious and lateral roots in r ice. Plant Breeding, (2006); 125: 198 -200.
- Qu Y, Mu P, Zhang H, Chen CY, Gao Y, Tian Y, Wen F, Li Z. Mapping QTLs of root morphological traits at different growth stages in rice. Genetica, (2008); 133: 187 – 200.
- Singh N, Choudhury RD, Tiwari G, Singh KA, Kumar S, Srinivasan K, Tyagi KR, Sharma DA, Singh KN, Singh R. Genetic diversity trend in Indian rice varieties: an analysis using SSR markers. BMC Genetics, (2016); 17:127 doi: 10.1186/s12863-016-0437-7. PMID: 27597653; PMCID: PMC5011800.
- Liu L, Wu Y, Wang Y, Samuels T. A high-density simple sequence repeat-based genetic linkage map of switchgrass. G3 (Bethesda, Md), (2012); 2: 357–370.
- Wu Y, He J, Li A, Fang N, He W, Dang L, Zeng G, Huang J, Bao Y, Zhang H. Population structure analysis and association mapping of blast resistance in indica rice (Oryza. sativa L.) landraces. Genetics of Molecular Research, (2016); 15: 1-11.
- Yao J, Wang L, Liu L, Zhao C, Zheng Y. Association mapping of agronomic traits on chromosome 2A of wheat. Genetica, (2009); 137: 67-75.
- Rana MM, Islam MA, Imran S, Ruban S, Hassan L. Genetic diversity analysis of NERICA lines and parents using SSR markers. Int. Journal of Plant and Soil Science, (2018); 23: 1-10.
- Ahmed S, Anik TR, Islam A, Uddin I, Haque MS. Screening of Some Rice (Oryza. sativa L.) Genotypes for Salinity Tolerance using Morphological and Molecular Markers. Biosciences Biotechnology Research Asia, (2019); 16: 377-390.
- Verma H, Borah J, Sarma R. Variability assessment for root and drought tolerance traits and genetic diversity analysis of rice germplasm using SSR markers. Scientific reports, (2019); 9:1-19.
- Shahriar M, Robin A, Begum S, Hoque A. Diversity analysis of some selected rice genotypes through SSR-based molecular markers. Journal of the Bangladesh Agricultural University (2014); 12: 307-311.
- Shah MS, Arif M, Aslam K, Shabir G, Thomson JM. Genetic diversity analysis of Pakistan rice (Oryza sativa) germplasm using multiplexed single nucleotide polymorphism markers. Genetic Resource and Crop Evolution, (2016); 63:1113–1126
- Anandan A, Anumalla M, Pradhan SK, Ali J. Population structure, diversity and trait association analysis in rice (Oryza. sativa L.) germplasm for early seedling vigor (ESV) using trait linked SSR markers. PloS One, (2016); 11: 1-9.
- Belamkar V, Selvaraj MG, Ayers JL, Payton PR, Puppala N, Burow MD. A first insight into population structure and linkage disequilibrium in the US peanut minicore collection. Genetica. (2011); 139:411
- Ahmed HG, Naeem M, Zeng Y, Rashid MAR, Ullah A, Saeed A. Genome-wide association mapping for high temperature tolerance in wheat through 90k SNP array using physiological and yield traits. PLoS ONE, (2022); 17(1): 1-11.
- Suvi WT, Shimelis H, Laing M, Mathew I, Shayanowako AIT. Assessment of the genetic diversity and population structure of rice genotypes using SSR markers. Acta Agriculturae Scandinavica, Section B— Soil & Plant Science, (2020); 70: 76-86.
- Courtois B, Frouin J, Greco R, Bruschi G, Droc G, Hamelin C, Ruiz M, Clément G, Evrard GC, Van S, Genetic diversity and population structure in a European collection of rice. Crop Science, (2012); 52:1663-1675.
- Ali LM, McClung AM, Jia MH, Kimball JA, McCouch SR, Georgia CE. A rice diversity panel evaluated for genetic and agro-morphological diversity between subpopulations and its geographic distribution. Crop Science, (2011); 51:2021-2035.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0
![]()


