

Full Length Research Article
Investigations on molecular determinants of durable molecular response in chronic myeloid leukemia patients
Zafar Iqbal
Adv. life sci., vol. 9, no. 1, pp. 67-74, May 2022
*- Corresponding Author: Zafar Iqbal (Email: drzafar.medgen@yahoo.com)
Authors' Affiliations
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Chronic myeloid leukemia (CML), a blood cancer, is caused by translocation between chromosomes 22 and 9 that gives rises to fusion oncogene BCR-ABL. In 20th century, CML was a deadly disease, but tyrosine kinase inhibitors (TKI) led to complete remission in over 80% CML patients. Nevertheless, TKIs are expensive, and discontinuation of treatment is required in patients with very stable treatment response. As no molecular markers of durable TKI response exist, this study was conducted to find out molecular determinants of durable response in CML patients treated with TKIs.
Methods: Peripheral blood and clinical data were collected from CML patients with durable treatment response, along with appropriate controls. DNA was extracted and whole exome sequencing (WES) carried out to screen novel genes mutated only in experimental groups and absent in control groups. Mutations were confirmed using Sanger sequencing. Data was analyzed using SPSS version 23.
Results: Although WES detected 10 genes mutated exclusively in CML patients with durable treatment response, Sanger sequencing could confirm mutations only in RAI1 gene (GC deletion at nucleotides 837-838, a frameshift mutation).
Conclusions: Our study shows that mutations in a novel gene (RAI1) are associated with durable response in CML patients. RAI1 gene is active throughout the body and controls functions of many genes involved in daily rhythms. Our studies provide first important insights into molecular factors associated with long-term treatment response in CML that can serve as novel biomarker to identify patients eligible for TKI cessation in many ongoing CML STOP-TKI trials.
Keywords: CML; TKI therapy; Durable response; Molecular biomarkers; STOP-TKI
Introduction![]()
Chronic myeloid leukemia (CML) is an acquired myeloproliferative neoplasm characterized by the presence of Philadelphia (Ph) chromosome resulting from reciprocal translocation between chromosomes 9 and 22, t (9;22) (q34; q11) [1]. This translocation involves the ABL1 gene on chromosome 9 and the BCR gene on chromosome 22 and formation of a BCR–ABL fusion gene [2]. This BCR–ABL is an oncogene which codes for 210 kDa oncoprotein with increased tyrosine kinase activity [3]. Imatinib mesylate (IM) is synthetic tyrosine kinase inhibitor (TKIs) designed as the first specific molecularly targeted drug to inhibit the BCR–ABL fusion protein in CML [4]. In last two decades, IM has been used as a gold standard drug for the treatment of newly diagnosed Ph chromosome positive CML patients [4]. Second and third generation tyrosine kinase inhibitors (TKIs) have further improved survival of CML patients and in recent era the life expectancy of chronic phase CML patients equals to general population at least in developed countries [5]. This led to the idea of cessation of TKI therapy in a subset of CML patients who have long-term response, also known as treatment-free remission.
Although TKI therapy is very effective, it is very expensive that makes initial clinical recommendations of prescribing TKIs for whole life impractical [6]. The expense of targeted therapies may hinder sustaining TKI-based treatment, not only in developing countries but even in developed countries [7,8]. Furthermore, due to high effectiveness of TKI therapy in recent era, achievement of the durable molecular response (log 4.5 reduction BCR-ABL1 transcripts) in about one third of CML patients makes them candidates for cessation of targeted therapy, termed as treatment-free remission (TFR) in recent years [8]. This led to the initiation of TFR trials in CML long-term TKI responders around the world that led to successful TKI-therapy cessation in a subset of CML patients with durable molecular response [9, 10]. Accordingly, recent CML clinical guidelines recommend TFR as a major objective of CML treatment in CML patients with durable or durable molecular response (DMR) for more than two years, in addition to other clinical parameters [11]. Nevertheless, very recent data suggests that about half of the patient subjected to cessation of TKI therapy experienced disease relapse [12]. Currently, clinical criteria to select CML patients with durable molecular response does not include any molecular biomarkers except molecular response monitoring, a parameter that has many levels and therefore has been adopted differently by different CML groups globally [8-12]. Historically, a lot of work has been done to find out biomarkers of inferior outcome of CML patients but there are very few studies to find out molecular biomarkers of superior outcome, specifically related to TKI treatment, in CML [9,10,13]. Therefore, it is the need of the hour to find out molecular biomarkers of durable molecular response in CML patients that can be utilized to identify patients that candidates for TFR in CML [12, 13]. Keeping that in view, this study was carried out to find novel genomic variants associated with long-term response to TKIs in CML patients by employing next-generation DNA sequencing.
Methods![]()
Patient Selection:
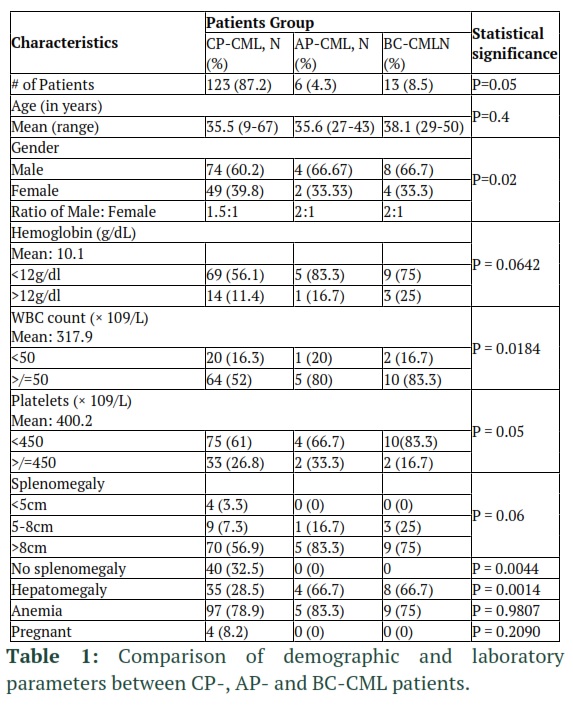
In this study, 142 Ph-chromosome positive CML patients were recruited who were receiving imatinib mesylate (IM) as first-line therapy and nilotinib (NI) as second-line treatment in case of IM resistance or non-responsiveness. These CML patients were recruited from Hayatabad Medical Complex (HMC) Peshawar, Khyber Pakhtunkhawa (KP) Pakistan, from January 2012 to Dec 2020. The age/sex matched healthy controls (n=10) were also included. Out of these patients, 123 were chronic-phase (CP-), 6 advanced phased (AP-) and 13 blast-crisis (BC-) CML patients. Patient characteristics are provided in Tables 1-2.
Per European LeukemiaNet 2020 recommendations, CP-CML patients with durable major molecular response (log 4.5 MMR for at least three consecutive years) were experimental group (60/142, 42.2%) while CML without durable molecular response (MMR less than log 4.5, n=63, 44.4%) were control group 1 (11). AP-CML (n=6, 4.2%) and BC-CML (n=13, 9.2%) were also included as additional control groups (control groups 2 and 3, respectively). Healthy controls (n=10) were included as control group 4. All response criteria were per European LeukemiaNet (ELN) guidelines 2013 (stated below) as treatment of patients started in Jan 2012 [6]. Study was approved by ethical review boards of participating centers. Consent was taken from the study subjects and the study was conducted in accordance with the Declaration of Helsinki.
Clinical Criteria:
Disease Phases: In the peripheral blood, CML-CP was characterized as the presence of less than 5% blast cells, 15 to 19% basophils, less than 30% blast and promyelocyte cells, and no evidence of blast cells in extramedullary sites [14]. Blasts in blood or marrow 15-29% or greater than or equal to 30% blasts and promyelocytes (with blasts count less than 30 percent) in blood or bone marrow were classified as CML-AP. The presence of equal to or higher than 30% blasts in blood or bone marrow, as well as blast cells in extramedullary sites other than the spleen, was used to assign BC-CML.
Response Criteria:
Complete hematological response (CHR) was defined as platelet count of less than 450 × 109/L, WBC count of less than 10 × 109/L, differential count without immature granulocytes and with less than 5% basophils, along with no extramedullary disease and all disease-related signs and symptoms have disappeared. Anything less than CHR was taken as partial hematological response (PHR). Complete cytogenetic response (CCyR) was defined as 0%-1% Ph+ cells out of ≥ 200 metaphases by cytogenetic analysis. Partial cytogenetic response (PCyR) was considered as 1-35%; Ph+ cells, minor cytogenetic response (mCyR) as 36–65% Ph+ cells and minimal cytogenetic response (miCyR) as 66–95% Ph+ cells while no cytogenetic response (nCyR) was taken as Ph+ cells equal to at least 95% (6) .
The ELN response to treatment was considered optimal at 3 months when Ph+ chromosomes were 35%, warning when Ph+ chromosomes were 36-95%, and failure when the percentage of Ph+ chromosomes was >95%. Similarly, optimal response at 6 months when Ph+ 0% and failure when Ph+ chromosomes were >95%, warning when Ph+ 1-35%, and failure when Ph+ >35%. Due to ELN requirements requiring molecular testing for interpretation, we were unable to measure the ELN treatment response at 12 months [6]. The revised ELN guidelines were used to define molecular responses. Major molecular response (MMR) was described as a bcr-abl /abl ratio cutoff of ≤0.1%, MR4 was considered as a ratio ≤0.01%, and MR4.5 as ratio of ≤ 0.0032% [6].
Survival definitions:
(i) From the start of IM therapy until the last follow-up date or the patients expired date, the OS (overall survival) was measured. (ii). PFS (progression-free survival) was measured from the start of first diagnosis to the recorded progression of CML to AP or BC, or until death, whichever came first. The Kaplan-Meier method [15] was used to assess the survival analysis. In those who developed intolerance because of therapy-related side effects, the IM dosage was decreased. If grade 3 or 4 toxicity was detected, the IM treatment was halted. IM therapy was start once again at a decreased dose when the toxicity diminished.
Study approval:
This study was approved by the institutional ethical review committees of all participating centers which comply with the Declaration of Helsinki [16]. A written informed consent was derived from all participants in the present study.
Patient Treatment:
Prior to treatment, patients were categorized into various risk categories, using clinical and laboratory data [6, 17]. Patients with CML-CP were given 400 mg of imatinib orally once a day. In IM non-responders or patients with suboptimal cytogenetic response (minor or minimal) after 6 months, the IM dose was increased to 600-800mg daily. Patients with new-onset AP or BC were given 600-800mg IM regular. Nilotinib was used as a first-line treatment in CP patients who developed resistance or intolerance to IM, and as a second-line treatment in AP and BC at a dose of 400mg twice a day, with doses increasing to 600mg to 800mg twice a day if response was low. The patient’s reaction to treatment was assessed clinically, and blood counts were checked every 4-8 weeks. At diagnosis, 6 months, 12 months, and yearly thereafter, aspirates from bone marrow were used for cytogenetics and molecular analysis using real-time quantitative PCR, and all definitions for molecular responses were adopted per international guidelines [18].
Sample Collection & DNA extraction
We utilized EDTA blood collection tubes to extract peripheral blood samples (3-5 mL) (BD Vacutainer Systems, Franklin Lakes, and N.J.). The QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) was used to remove genomic DNA (gDNA) from the entire blood of advanced-stage CML patients [19]. The DNA was quantified with NanoDrop Spectrophotometer (NanoDrop Technologies, Inc., USA) and diluted into 70-80ng/l aliquots for whole exome sequencing (WES). The rest of the DNA was diluted to 40ng/l for mutation validation and later use on Sanger sequencing. DNA was kept in a -80°C freezer until it was time to test it.
Whole Exome Sequencing (WES)
WES was carried out at Centre for Genetics and Inherited Diseases, College of Medicine, Taibah University, Medina Saudi Arabia. For library preparation and exome enrichment, we used the SureSelectXT V6-Post Capture Exome package (SureSelect, Agilent). Exonic and intron flanking regions were enriched using Illumina Paired-End Multiplexed Sequencing (SureSelectXT2) according to manufacturer protocol. In a brief, 50ng of DNA was fragmented and tagmentation was performed. Purification and amplification followed the DNA tagmentation process. The amplified DNA libraries were purified with magnetic beads, and target regions were captured with oligos (entire exome), followed by PCR amplification of the enriched libraries. The enriched libraries were quantified using a Qubit fluorometer, and the distribution of library size was calculated using an Agilent Bioanalyzer. Finally, the quantified DNA libraries were loaded onto a flow cell for cluster generation and whole exome sequencing using an Illumina NextSeq500 computer [20].
Data Analysis for Exome sequencing
BCL2FASTQ program was used to convert the WES output BCL files to FASTQ files. BWA aligner was used to align the FASTQ files to the human genome (GRCh37/hg19) using the BWA-MEM algorithm. The GATK (Genome Analysis Tool Kit) was used to name variants. Illumina Variant Studio was used to annotate and filter genomic variants [20].
Strategy for variant analysis
To discover a common biomarker of CML progression, we first focused on DNA repair genes that were mutated in all advanced phase CML patients. WES annotated Excel file and was processed in the following manner: first, all synonymous and intron variants were removed. Second, “rare variants calling” was performed, which was described as a filtration strategy for all variants with less than 70% of allele frequencies. Furthermore, all Tolerant (T) and Benign (B) Variants that had a known prediction were removed. We considered multiple B or T to be B if the frequency of B was 70% or T if the frequency of T was 70%, and all TBB combinations were eliminated. Finally, WES data were examined for driver mutations, which are gene variants that are not found in CP-CML patients but are found in AP-BC CML patients, suggesting that these variants may be involved in disease progression.
Variants validation by Sanger Sequencing
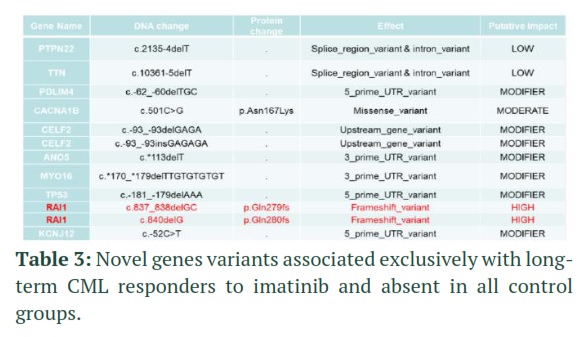
To confirm the WES-identified variants in PTPN22, TTN, PDLIM4, CACNA1B, CELF2, ANO5, MYO16, TP53, RAI1 and KCNJ12, Sanger sequencing [21] was used. The University of California Santa Cruz genome database browser (http://genome.ucsc.edu/cgibin/hgGateway) was used to obtain specific genomic primers for the variants in detected genes. For sample preparation, a Sanger sequencer, ABI Prism 3730 Genetic Analyzer (Applied Biosystems, California, USA), and ABI PRISM Big Dye Terminator Cycle Sequencing Ready Reaction kits were used (V.3). On CML patients, whole-exome sequencing was performed. We confirmed RAI1 gene since it is associated with good response to imatinib. The RAI1 gene, which is active throughout the body and regulates the functions of many genes involved in daily rhythms, had different frameshift mutations.
Statistical Analysis of patient data
Based on the normality test, we presented absolute numbers and percentages for categorical variables, as well as mean and median with an adequate measure of variance for continuous variables. Association between two groups for categorical data were compared using Chi-Square or Fisher’s exact test. For continuous data, the two-sample independent test or the Mann Whitney U test was used, depending on the normality hypothesis. For ≥3 groups, analysis of variance (ANOVA) or Kruskal-Wallis test was utilized. All statistical tests were performed using [SAS/STAT] software version 9.4 (SAS Institute Inc., Cary, NC, USA.) and for statistical computing, R foundation was used (Vienna, Austria). The Sokal risk score was determined according to the literature description [17].
Results![]()
Overall, 142 CML patients were included in the study, with mean age of 35.5 (range: 9-67) year and male to female ratio of 1.6:1. The mean hemoglobin, WBC count and platelet count were 10.1, 317.9 and 400.2, respectively. CP-, AP- and BC-CML phase patients were 123 (86.6%), 6 (4.2%) and 13 (9.2%), respectively. The mean age of CP, AP, and BC CML patients was 35.5, 35.6 and 38.1 years, respectively (p=0.42). Overall, male was more affected than females at all disease phases with male to female ratio of 1.5:1, 2:1 and 1.6:1 for CP-, AP- and BC-CML, respectively (p=0.02). Furthermore, CP-CML were significant different from advanced phase patients with respect to male to female ratio, hemoglobin level, WBC count, platelet count and hepatomegaly (Table 1).
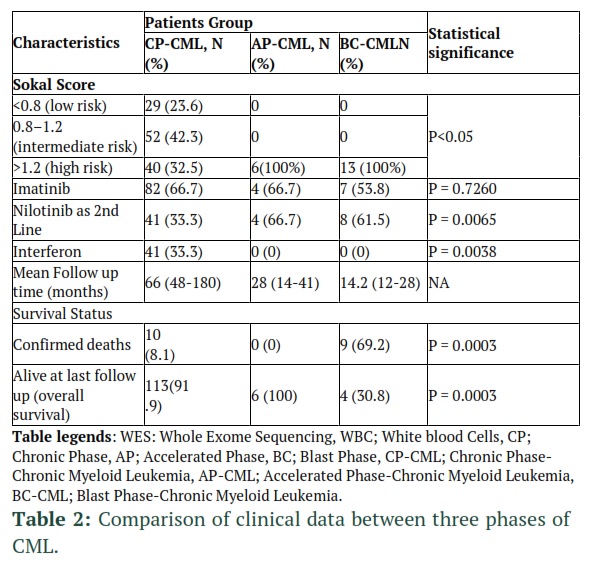
Overall, 66.7% (82), 4 (66.7%) and 7 (53.8%) of CP-, AP- and BC-CML patients received standard imatinib dose, respectively. About 33.3% (41) CP-CML switched from interferon to imatinib. All patients resistant or intolerant to imatinib were switched to standard dose nilotinib. Other second generation and third generation TKIs were not available. Mean follow-up of CP-, AP- and BC-CML was 66, 28 and 14.2 months, respectively. Among CP-CML patients, 60 (42.2%) had durable molecular response (log 4.5 MMR durable for at least three consecutive years) while 63 (44.4%) CP-CML patients did not quality criteria for DMR. Therefore, our CML patients were grouped into CP-CML with DMR (experimental group), CP-CML without DMR (control group 1), AP-CML (control group 2) and BC-CML (control group 4) patients. It is important to mention that as all CP-CML patients in experimental group and control group 1 had at least MMR, both groups had no statistical difference with respect to clinical features, except durable molecular response.
Exome sequencing
Initial screening for mutations for genes related to durable response
In initial WES screening, mutations were detected in PTPN22, TTN, PDLIM4, CACNA1B, CELF2, ANO5, MYO16, TP53, RAI1 and KCNJ12 for CP-CML with durable MMR (experimental group). Mutations in these genes were not detected in any of control groups as well as in healthy controls (Table 2)
Mutation validation by Sanger Sequencing
Sanger sequencing was employed to confirm mutations in PTPN22, TTN, PDLIM4, CACNA1B, CELF2, ANO5, MYO16, TP53, RAI1 and KCNJ12 in all patient groups. We could confirm RAI1 gene mutation (GC deletion at nucleotides 837-838) in our experimental group (CP-CML patients with durable MMR) but in none of other control groups. Bioinformatics analysis revealed that this two-nucleotide deletion leads to frameshift mutation at amino-acid 279 (Gln279fs) which leads to change in all amino-acids downstream of Gln279 in RAI1 protein (Table 3). It shows that this RAI1 mutation is associated with durable MMR in CML patients and could be potential new biomarker for durable molecular response in CML patients treated with tyrosine kinase inhibitors.
Figures & Tables
Discussion![]()
CML was associated with younger age in our patient groups. We found mean age of CP, AP, and BC CML patients to be 33.5, 35.6 and 38.1 years in our studies, which is in accordance with various other studies [23]. Bansal et al. reported median age of 38 years in newly diagnosed Indian CML patients [24]. Two previous studies published from Pakistan have reported median ages of 38 and 37 years for CML patients [25]. In contrast to our study a study conducted in Atlanta reported the median age for CML patients was 74.8 years [27].
In our studies, 42.2% of CP-CML patients had durable molecular response (MMR durable for at least three consecutive years). Similarly, a study conducted in UK showed that 35% imatinib-treated CML patients showed durable MMR [26]. Another study carried out in Pakistan reported 86% MMR and 39% durable molecular response [25].
The goal of current management of patients with chronic phase chronic myeloid leukemia (CML) is to reach treatment-free remission with durable deep molecular remission (DMR) being the prerequisite. Therefore, Second-generation tyrosine kinase inhibitors can induce deeper and faster remission than imatinib. Nilotinib was noted to provide the highest rate of MMR among other TKIs [28]. a retrospective observational study conducted in — found that patients with chronic-phase CML who had been prescribed a first line imatinib. A durable molecular response was observed in 50% and 29% of the patients, however more durable molecular responses were observed in patients who receive a second-generation TKI second line 77% and 44% of evaluable patients with ≥13 months follow-up [29].
Finding CML patients who achieve DMR has clinical significance as these patients are candidates for TFR and discontinuation of TKI therapy which is focus of many ongoing clinical trials [30]. Unfortunately, no reliable biomarkers of DMR and TFR are currently available that can early identify such CML patient groups [31]. In our studies, exome sequencing of CP-CML patients’ groups with DMR found RAI gene mutation (GC deletion at nucleotides 837-838, corresponding to frameshift mutation at amino-acid 279/ Gln279fs). This mutation was detected in all DMR CP-CML patients but in none of non-DMR CP-CML, AP-CML, BC-CML or healthy controls. RAI gene has never been reported in any type of leukemia or other related diseases [32].
Retinoic acid-inducible or induced gene 1 (RAI-1) was firstly identified as a main regulator of neuronal and glial differentiation induced during a mouse model of embryonal carcinoma cell line after treatment with high doses of retinoic acid [32]. At the transcriptional level, retinoids regulate gene expression in a variety of cells and tissues. Downstream regulatory molecules such as enzymes, transcription factors, cytokines, and cytokine receptors are all transcriptionally regulated by retinoic acid. The role of retinoid signaling pathways in the regulation of synaptic plasticity and learning has been demonstrated in animal models. (ref11). The RAI1 gene provides instructions for making a protein that is active in cells throughout the body, particularly nerve cells (neurons) in the brain. Located in the nucleus of the cell, the RAI1 protein helps control the activity (expression) of certain genes [32]. Most of the genes regulated by RAI1 have not been identified. Mutations causing decreased amount of RAI1 gene are responsible for a phenotype like 17p11.2 deletion syndrome [33]. However, studies suggest that this protein controls the expression of several genes involved in daily (circadian) rhythms, such as the sleep-wake cycle. The RAI1 protein also appears to play a role in development of the brain and of bones in the head and face (craniofacial bones) [32].
RAI1 is a dosage-sensitive gene. Several studies have indicated that the chromosomal segment of RAI1 gene is critically involved in both SMS and Potocki–Lupski syndrome with deletions and duplications, respectively [34]. Murine models of SMS, deletions of the area containing RAI1 or focused on targeted gene inactivation, cause the vast majority of the clinical features seen in the SMS patients (craniofacial anomalies, obesity, unusual circadian rhythm, and seizures) [35]. However, murine models with duplication incorporating RAI1 or with additional duplicates of RAI1 showed entirely restricting aggregates in hyperphagia, body weight, percent of muscle to fat ratio, anxiety, social and behavioral conduct, and hyperactivity [36].
A recent study identified two SNPs (rs9907986 and rs4925102) in the 5′-upstream region (5′-UTR) as putative regulatory elements to investigate the possible involvement of RAI1 in the pathogenesis of neuropsychiatric disorders [37, 38]. These two SNPs, which fall within the binding sites for the transcription factors DEAF1 and the retinoic acid RXRα-RARα, account for 30–40% of the variance in RAI1 mRNA expression in the prefrontal and temporal cortex, respectively. A recent study found a mutation in DEAF1 in a patient with clinical features like the normal SMS but negative for both the 17p11.2 deletion and RAI1 mutations, further supporting the function of DEAF1 in controlling RAI1 expression identified by exome sequencing [39].
Genetic variations are associated with clinical responses to Imatinib Mesylate. The human organic cation transporter 1 (OCT1; SLC22A1) has been reported to be the main influx transporter involved in IM uptake into CML cells. IM response may be influenced by genetic variations and/or changes in hOCT1 expression. When compared to the IM-responder group, the hOCT1 gene was significantly downregulated in the samples of the IM-resistant group (p = 0.0211), according to a study of 69 CML patients [6]. Moreover, IM uptake is mediated by the hOCT1 protein generated by the solute carrier 22 gene (SLC22A1). A study has investigated the impact of few single-nucleotide polymorphisms (SNPs) of SLC22A1 on mediating resistance and/or good response to IM among 278 Malaysian CML patients. Results showed as compared to the IM good response group, allelic frequencies of heterozygous (CG) and homozygous variant (GG) genotypes of SLC22A1 C480G were significantly higher in the IM-resistant group (41.8% versus 30.3% and 10.9% versus 4.5% with P values of 0.047 and 0.048, respectively) [40].
In nearly all cases, patients who relapsed in these trials remained sensitive to TKIs and regained MMR upon re-treatment. Based on these findings, Imatinib is highly effective in most patients with CML-CP; patients who respond are likely to live substantially longer than those treated with earlier therapies [5]. Achieving CCyR correlated with PFS and overall survival but achieving MMR had no further predictive value. Only restricted CML patients will currently maintain a TFR. While this target appeals to young patients and those without other comorbidities, we agree that it will eventually become the therapy goal for all CML patients if we can find ways to increase the rate of effective discontinuation with a minimum of side effects. As a result, efforts to incorporate TFR through biological and clinical research programs must also consider logistic procedures that are simpler than those currently recommended, in order to expand the TFR option to a larger population of CML patients around the world.
The author declares that he has no conflict of interest.
References
- Frazer R, Irvine AE, McMullin MF. Chronic Myeloid Leukaemia in the 21st Century. Ulster Medical Journal, (2007); 76(1): 8-17.
- Kang ZJ, Liu YF, Xu LZ, Long ZJ, Huang D, Yang Y, et al. The Philadelphia chromosome in leukemogenesis. Chinese Journal of Cancer, (2016); 35:48.
- Togasaki E, Takeda J, Yoshida K, Shiozawa Y, Takeuchi M, Oshima M, et al. Frequent somatic mutations in epigenetic regulators in newly diagnosed chronic myeloid leukemia. Blood Cancer Journal, (2017); 7(4): e559.
- Pophali PA, Patnaik MM. The Role of New Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Cancer Journal. (2016); 22(1): 40-50.
- Haznedaroglu IC. Monitoring the Response to Tyrosine Kinase Inhibitor (TKI) Treatment in Chronic Myeloid Leukemia (CML). Mediterranean Journal of Hematology and Infectious Diseases. (2014); 6(1): e2014009.
- Ben Hassine I, Gharbi H, Soltani I, Teber M, Farrah A, Ben Hadj Othman H, et al. hOCT1 gene expression predict for optimal response to Imatinib in Tunisian patients with chronic myeloid leukemia. Cancer Chemotherapy and Pharmacology, (2017) ; 79(4): 737-745.
- Chen Q, Jain N, Ayer T, Wierda WG, Flowers CR, O'Brien SM, et al. Economic Burden of Chronic Lymphocytic Leukemia in the Era of Oral Targeted Therapies in the United States. Journal of Clinical Oncology, (2017); 35(2): 166-174.
- D Irani YD, Hughes A, Clarson J, Kok CH, Shanmuganathan N, White DL, et al. Successful treatment-free remission in chronic myeloid leukaemia and its association with reduced immune suppressors and increased natural killer cells. British Journal of Haematology, (2020); 191(3): 433-441.
- D Makhtar SM, Husin A, Baba AA, Ankathil R. Genetic variations in influx transporter gene SLC22A1 are associated with clinical responses to imatinib mesylate among Malaysian chronic myeloid leukaemia patients. Journal of Genetics, (2018); 97(4): 835-842.
- D Shih YT, Cortes JE, Kantarjian HM. Treatment value of second-generation BCR-ABL1 tyrosine kinase inhibitors compared with imatinib to achieve treatment-free remission in patients with chronic myeloid leukaemia: a modelling study. Lancet Haematology, (2019); 6(8): e398-e408.
- Saußele S, Richter J, Hochhaus A, Mahon FX. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia, (2016); 30(8): 1638-47.
- Gong Z, Zheng L, Tang Z, Chen Z, Wang W, Bai S, et al. Role of complexity of variant Philadelphia chromosome in chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Annals of Hematology, (2017); 96(3): 501-504.
- Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia. Blood, (2013); 122(6): 872-884
- Cortes JE, Talpaz M, O'Brien S, Faderl S, Garcia-Manero G, Ferrajoli A, et al. Staging of chronic myeloid leukemia in the imatinib era: An evaluation of the World Health Organization proposal. Cancer, (2006); 106(6): 1306-1315.
- Kaplan E and Meier P. Nonparanietric estimation from incomplete observations. Journal of the American Statistical Association, (1958); 53(282): 457-481.
- Goodyear MD, Krleza-Jeric K, Lemmens T. The Declaration of Helsinki. The BMJ, (2007); 335(7621): 624-5.
- Sokal JE, Cox EB, Baccarani M, Tura S, Gomez GA, Robertson JE, et al. Prognostic discrimination in "good-risk" chronic granulocytic leukemia. Blood, (1984); 63(4): 789-799.
- Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European Leukemia Net. Blood, (2006); 108(6): 1809-1820.
- Cumbo C, Impera L, Minervini CF, Orsini P, Anelli L, Zagaria A, et al. Genomic BCR-ABL1 breakpoint characterization by a multi-strategy approach for "personalized monitoring" of residual disease in chronic myeloid leukemia patients. Oncotarget, (2018); 9(13): 10978-10986.
- Al-Asiri S, Basit S, Wood-Trageser MA, Yatsenko SA, Jeffries EP, Surti U, et al. Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. Journal of Clinical Investigation, (2015); 125(1): 258-262.
- Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR et al. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. The Journal of Molecular Diagnostics, (2010); 4: 425-432.
- Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutations in RAI1 associated with Smith-Magenis syndrome. Nature Genetics, (2003); 33(4): 466-8.
- Kuntegowdanahalli LC, Kanakasetty GB, Thanky AH, Dasappa L, Jacob LA, Mallekavu SB, et al. Prognostic and predictive implications of Sokal, Euro and EUTOS scores in chronic myeloid leukaemia in the imatinib era-experience from a tertiary oncology centre in Southern India. Ecancermedicalscience, (2016); 10: 679.
- Bansal S, Prabhash K, Parikh P. Chronic myeloid leukemia data from India. Indian Journal of Medical and Paediatric Oncology, (2013); 34(3): 154–158.
- Zaidi U, Kaleem B, Borhany M, Maqsood S, Fatima N, Sufaida G, et al. Early and durable deep molecular response achieved with nilotinib in high Sokal risk chronic myeloid leukemia patients. Cancer Management Research, (2019); 11: 1493-1502.
- de Lavallade H, Apperley JF, Khorashad JS, Milojkovic D, Reid AG, Bua M, et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: incidence of durable responses in an intention-to-treat analysis. Journal of Clinical Oncology, (2008); 26(20): 3358-63.
- Rousselot P, Cony-Makhoul P, Nicolini F, Mahon FX, Berthou C, Réa D, et al. Long-term safety and efficacy of imatinib mesylate (Gleevec®) in elderly patients with chronic phase chronic myelogenous leukemia: results of the AFR04 study. American Journal of Hematology, (2013); 88(1): 1-4.
- Heibl S, Buxhofer-Ausch V, Schmidt S, Webersinke G, Lion T, Piringer G, et al. A phase 1 study to evaluate the feasibility and efficacy of the addition of ropeginterferon alpha-2b to imatinib treatment in patients with chronic phase chronic myeloid leukemia (CML) not achieving a deep molecular response (molecular remission 4.5)-AGMT_CML 1. Hematological Oncology, (2020); 38(5): 792-798.
- Milojkovic D, Cross NCP, Ali S, Byrne J, Campbell G, Dignan FL, et al. Real-world tyrosine kinase inhibitor treatment pathways, monitoring patterns and responses in patients with chronic myeloid leukaemia in the United Kingdom: the UK TARGET CML study. British Journal of Haematology, (2021); 192(1): 62-74
- Cortes J, Rea D, Lipton JH. Treatment-free remission with first- and second-generation tyrosine kinase inhibitors. American Journal of Hematology, (2019); 94(3): 346-357.
- Irani YD, Hughes A, Clarson J, Kok CH, Shanmuganathan N, White DL, et al. Successful treatment-free remission in chronic myeloid leukaemia and its association with reduced immune suppressors and increased natural killer cells. British Journal of Haematology, (2020); 191(3): 433-441.
- Fragoso YD, Stoney PN, Shearer KD, Sementilli A, Nanescu SE, Sementilli P, et al. Expression in the human brain of retinoic acid induced 1, a protein associated with neurobehavioural disorders. Brain Structure and Function, (2015); 220(2): 1195-203.
- Vilboux T, Ciccone C, Blancato JK, Cox GF, Deshpande C, Introne WJ, et al. Molecular analysis of the Retinoic Acid Induced 1 gene (RAI1) in patients with suspected Smith-Magenis syndrome without the 17p11.2 deletion. PLoS One. (20110; 6(8): e22861.
- Ricard G, Molina J, Chrast J, Gu W, Gheldof N, Pradervand S, et al. Phenotypic consequences of copy number variation: insights from Smith-Magenis and Potocki-Lupski syndrome mouse models. PLoS Biology, (2010); 8(11): e1000543.
- Yan J, Bi W, Lupski JR. Penetrance of craniofacial anomalies in mouse models of Smith-Magenis syndrome is modified by genomic sequence surrounding Rai1: not all null alleles are alike. American Journal of Human Genetics, (2007); 80(3): 518–525
- Bi W, Yan J, Shi X, Yuva-Paylor LA, Antalffy BA, Goldman A, et al. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Human Molecular Genetics, (2007); 16(15): 1802-13.
- Chen L, Tao Y, Song F, Yuan X, Wang J, Saffen D. Evidence for genetic regulation of mRNA expression of the dosage-sensitive gene retinoic acid induced-1 (RAI1) in human brain. Scientific Reports, (2016); 6: 19010.
- Vulto-van Silfhout AT, Rajamanickam S, Jensik PJ, Vergult S, de Rocker N, Newhall KJ, et al. Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. American Journal of Human Genetics, (2014); 94(5): 649-61.
- Berger SI, Ciccone C, Simon KL, Malicdan MC, Vilboux T, Billington C, et al. Exome analysis of Smith-Magenis-like syndrome cohort identifies de novo likely pathogenic variants. Human Genetics, (2017); 136(4): 409–420.
- Makhtar SM, Husin A, Baba AA, Ankathil R. Genetic variations in influx transporter gene SLC22A1 are associated with clinical responses to imatinib mesylate among Malaysian chronic myeloid leukaemia patients. Journal of Genetics, (2018); 97(4): 835-842.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0


