

Review Article
Contribution of GJB2 gene mutations to hearing loss in Pakistani population – A Narrative Review
Ejaz Ali*, Nageen Hussain
Adv. life sci., vol. 8, no. 3, pp. 217-220, July 2021
*- Corresponding Authors: Ejaz Ali (Email: ejazali067@gmail.com)
Authors' Affiliations
Abstract![]()
Introduction
Methods
Discussion
Conclusion
References
Abstract
Pakistan has a unique population for the study of recessive genetic diseases due to a higher consanguinity rate. Hearing impairment is the loss of hearing normal sounds, and it is a common sensory disorder that affects more than 466 million people worldwide. Immuno-genetic and other environmental factors like loud noises, drug usage, and viral infections are the causes of hearing loss. Hearing loss is categorized into a syndromic hearing loss (70%) and non-syndromic hearing loss (30%). GJB2 mutations are one of the main causes of hearing loss in different populations, including Pakistan. The GJB2 gene encodes a gap junction protein involved in the homeostasis of the inner ear through the recycling of potassium ions. The prevalence of GJB2 mutation in the Pakistani population varies from 6.1 to 9.2%. The most common mutations found in the Pakistani population are 71G>A (p.(Trp24*), 231G > A (p. Trp77*), c.35delG (p. Gly11Leufs24*),c.355G>T (p. Glu119*) 457G > A (p.Val153Ile), 598G > A (p.Gly200Arg), 439G > A (p.Glu147Lys), c.377_378insATGCGGA (p.Arg127Cysfs*85). c.1055C>T (p. Pro352Leu), c.6202A>C p.(Thr2068Pro), c.2496_2496delC p.(Tyr832*) and c.355G>T p.(Glu119*).
Keywords: GJB2 gene; Connexin 26 protein; DFNB1 locus; Hearing loss
Introduction![]()
Hearing loss (HL) or hearing impairment is the loss of hearing normal sounds [1]. It is one of the most common diseases that affect 466 million people worldwide and 1in 500-1000 newborns [2]. HL is classified based on different parameters such as onset, severity, and other associated diseases. Thus, HL is caused by environmental (high-intensity sounds, viral infections, and drug usage) and immuno-genetic factors. The genetic contribution to HL is a great challenge. Genetic and immunological factors cause almost 50 % of the HL. [3, 4].
Inheritance of HL can be recessive, dominant, X-linked, and mitochondrial [5]. HL due to genetic factors is classified into two categories, which are syndromic hearing loss (SHL), representing 20-30%, and non-syndromic hearing loss (NSHL), representing 70-80% cases [6]. More than 400 different syndromes are associated with SHL, which is categorized based on the mode of inheritance to the next generation, such as the maternal mode of inheritance due to mitochondrial gene mutations [7]. NSHL may follow an autosomal recessive (ARNSHL) or autosomal dominant (ADNSHL) mode of inheritance [8]. The gene locus for NSHL is known as DFN (DeaFNess). The locus for a gene that inherits in an autosomal dominant pattern is DFNA, autosomal recessive pattern as DFNB, and X-linked pattern as DFN [7]. It is estimated that more than 200 genes are responsible for HL; however, only 121 genes have been identified [9]. One of the leading genetic causes is mutations in the GJB2 gene, which encodes a gap junction connexin 26 protein, involved in the inner ear's homeostasis ear through the recycling of potassium ions [10].
Methods![]()
Literature Search Strategy and Selection Criteria
A systematic search was carried out from Google Scholar, NCBI, and Google Web Browsers through key terms GJB2, non-syndromic hearing loss, hearing impairment, and deafness. The literature found was further screen by inclusion and exclusion criteria. Recent research papers were selected. Research articles before 2005 were excluded. More than 20 articles were selected.
Discussion![]()
Mechanism of hearing loss
The cochlea comprises three fluid-filled compartments known as scala tympani, scala media, and scala vestibule. The scala tympani and scala vestibule contain a fluid (perilymph) with a high concentration of Na+ and low concentration of K+; whereas, scala media is filled with fluid (endolymph) which contain high concentrations of K+ and low concentration of Na+. The endolymph in scala media also includes a high positive endocochlear potential equal to 110 to 120 mV. This potential drives the K+ ion present in endolymph through the mechano-transduction in the hair's cells. Expelled out of K+ ions in the extracellular space is necessary to restore the cell polarization. This extra expelled K+ around hair cells needs to be removed to avoid K+ ion toxicity and maintain hair cell function. According to the K+ recycling hypothesis, these K+ ions are sunken by neighboring cells and transported back to endolymph through gap junction. [11]During auditory transduction in the cochlea of the inner ear cochlea, GJB2 (Gap junction beta-2 protein) maintains potassium homeostasis [5]. The six connexins 26 (Cx26) molecules assemble and form a single connexon in each cochlea cell, where Cx26 is mostly expressed. A gap junction between two connexons of the neighboring cochlea cells is formed, which serves as a path to maintain the K+ ion concentration balance between the different fluid compartments of the inner ear. Mutations in the GJB2 gene disrupt the communication channels between these cells, cause an imbalance of the potassium ion exchange and affect hearing function [12].
GJB2 gene involvement in hearing loss
GJB2 gene is one of the most common causes of non-syndromic hearing loss, and almost 300 different variants have been reported. The most common hereditary type of hearing impairment is ARNSHL, and more than 700 different causative mutations have been identified in over 80 DFNB loci. DFNB1 locus on chromosome 13q12.11 contains the most common ARNSHL gene (GJB2) with a single coding-exon, and more than 50% ARNSHL is linked with GJB2 mutations. More than 100 GJB2 mutations responsible for ARNSHL have been documented so far [13].
Prevalence of GJB2 gene mutation in Pakistani population
The prevalence of GJB2 mutations is different in different areas. According to the WHO, 2012, the prevalence rate of HL in Pakistan was 2.4% higher than the world's prevalence of 1.7% [13]. Compared to other populations, it is one of the most common mutations in Pakistan[13]. Consanguineous marriages increase the rate of recessive genetic disorders. High consanguinity in any population may lead to genetic diseases like HL. In Pakistan, consanguineous marriages are high; therefore, the population is unique to study recessive genetic disorders such as HL. The prevalence of profound bilateral HL in Pakistan is 1.6 per 1000 population, and 70% of the disorder arises in consanguineous families [7]. The studies conducted on the Pakistani population revealed a variable prevalence of GJB2 ranging from 4.2% to 53.33% [14-16].
In the Pakistani population, the contribution of GJB2 to NSHL is varied from 4.2 to 53.33%. In 2005, Santos et al. showed that only 6.1% of the studied families in Pakistan had GJB2 linked HL [17]. The most common mutations found in this report were p.Trp* and p.Trp77*, p.Arg2His, and p.Leu90Pro mutations were also detected. The 70% studied families belonged to the Punjab province, and another 30% belonged to other Pakistan provinces. In 2013, Bukhari et al. found a low prevalence (4.28%) of GJB2 mutation in the Hazara division, KPK province, compared to other provinces. Only three mutations, p.Trp24*, p. Ile35Ser, and p.Gly11Leufs24*, were found after sequencing the DNAs of 70 deaf patients (18). However, in 2014, Anjum et al. showed that the frequency of GJB2 mutation is higher (50%) in deaf patients from different areas of Pakistan [18]. The reported mutations were Trp24*, Gly12Valfs*2, Gln124*, Trp77*, Val153Ile, and Thr8Met. Similarly, in 2014 Shafique et al. revealed that the prevalence of GJB2 gene mutations in deaf patients of Punjab province was 53% [14]. The most common mutations were p.Trp24* and p.Trp77*. Other identified mutations were p.Gly200Arg p.Gly11Leufs24*, p.Glu147Lys, and p.R127Cfs*85. In 2017, Wang et al. revealed only mutation Gly11Leufs24* (35 del G). Similarly, in 2017, Shaikh et al. studied GJB2 mutations in a population of Sindh province, and prevalence was 6 % (9/150). The mutations found during the study were c.71G> A (p. Trp24*), c.231G> A (p.Trp77*), C35delG (Gly11Leufs24*), c.233delC (p.Leu79Cysfs3*), c.310del14 (p.Leu79Cysfs3*) , c.380G>A (p.Arg127His) as mention in table 01 and table 02. In 2019, mutations found in GJB2, were c.1055C>T (p.Pro352Leu), c.6202A>C p.(Thr2068Pro), c.2496_2496delC p.(Tyr832*) and c.355G>T p.(Glu119*) [13].
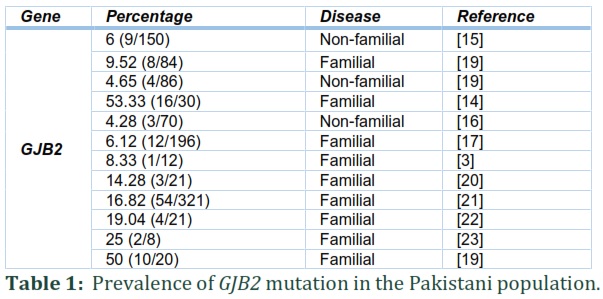
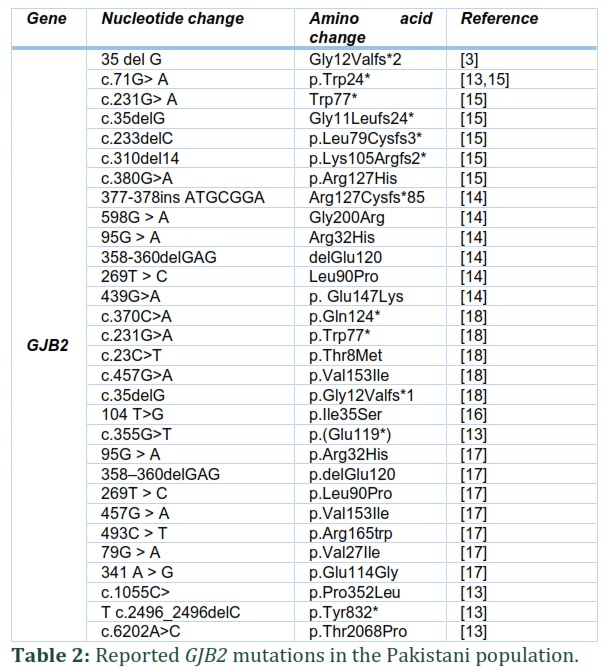
This narrative review concluded that non-syndromic hearing loss NSHL is caused by a mutation in more than 100 genes. The prevalence of hearing loss due to mutation in GJB2 varies from 4.2% to 53.33%. One of the most common causes of NSHL in the Pakistani population is a mutation in the GJB2 gene. The prevalence of GJB2 mutation in Pakistan is higher than the world's prevalence due to a higher rate of consanguinity. The most common mutations were p. Trp24* and p. Trp77*. Some recent mutations of GJB2 found are c.1055C>T (p. Pro352Leu), c.6202A>C p. Thr2068Pro, c.2496_2496delC p.(Tyr832*) and c.355G>T p.(Glu119*). The rate of NSHL can be reduced by avoiding consanguineous marriages. People should be educated about the consequences of consanguinity. This narrative review will be an excellent addition to the literature to understand hearing loss.
Both authors have equally contributed to the manuscript. Ejaz Ali wrote the manuscript, and Nageen Hussain edited and reviewed the manuscript.
The authors certify that they have no conflict of interest to declare in the subject matter or materials discussed in this manuscript.
References ![]()
- Naz S, Imtiaz A, Mujtaba G, Maqsood A, Bashir R, et al. Genetic causes of moderate to severe hearing loss point to modifiers. Clinical genetics, (2017); 91(4): 589-598.
- Wang R, Han S, Khan A, Zhang X. Molecular analysis of twelve Pakistani families with non-syndromic or syndromic hearing loss. Genetic testing and molecular biomarkers, (2017); 21(5): 316-321.
- Dror AA, Avraham KB. Hearing impairment: a panoply of genes and functions. Neuron, (2010); 68(2): 293-308.
- Fang Y, Gu M, Wang C, Suo F, Wang G, et al. GJB2 as well as SLC26A4 gene mutations are prominent causes for congenital deafness. Cell biochemistry and biophysics, (2015); 73(1): 41-44.
- Subaşıoğlu A, Duman D, Sırmacı A, Bademci G, Carkıt F, et al. Research of genetic bases of hereditary non-syndromic hearing loss. Turkish Archives of Pediatrics/Türk Pediatri Arşivi, (2017); 52(3): 122.
- Ali G. Genetic deafness in Pakistani population. J Pak Med Assoc, (2010); 60(6): 418-419.
- Atik T, Onay H, Aykut A, Bademci G, Kirazli T, et al. Comprehensive analysis of deafness genes in families with autosomal recessive non-syndromic hearing loss. PLoS One, (2015); 10(11): e0142154.
- Singh PK, Ghosh M, Sharma S, Shastri S, Gupta N, et al. Identification of a novel homozygous mutation in transmembrane channel like 1 (TMC1) gene, one of the second-tier hearing loss genes after GJB2 in India. The Indian journal of medical research, (2017); 145(4): 492.
- Koohiyan M, Hashemzadeh-Chaleshtori M, Salehi M, Abtahi H, Reiisi S, et al. GJB2 mutations causing autosomal recessive non-syndromic hearing loss (ARNSHL) in two Iranian populations: Report of two novel variants. International Journal of Pediatric Otorhinolaryngology, (2018); 107: 121-126.
- Zhao H-B. Hypothesis of K+-recycling defect is not a primary deafness mechanism for Cx26 (GJB2) deficiency. Frontiers in molecular neuroscience, (2017); 10162.
- Jaradat SA, Jubran B, Alzoubi F, Backe PH, Bader HM, et al. Molecular Analysis of the GJB2 Gene in Iraqi Patients with Sensorineural Non-Syndromic Hearing Loss. (2016). (2016); 171(3984): 1-11.
- Pavithra A, Chandru J, Jeffrey JM, Karthikeyen N, Srisailapathy CS. Rare compound heterozygosity involving dominant and recessive mutations of GJB2 gene in an assortative mating hearing impaired Indian family. European Archives of Oto-Rhino-Laryngology, (2017); 274(1): 119-125.
- Richard EM, Santos‐Cortez RLP, Faridi R, Rehman AU, Lee K, et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Human mutation, (2019); 40(1): 53-72.
- Shafique S, Siddiqi S, Schraders M, Oostrik J, Ayub H, et al. Genetic spectrum of autosomal recessive non-syndromic hearing loss in Pakistani families. PloS one, (2014); 9(6): e100146.
- Shaikh H, Waryah AM, Narsani AK, Iqbal M, Shahzad M, et al. Genetic Testing of Non-familial Deaf Patients for CIB2 and GJB2 Mutations: Phenotype and Genetic Counselling. Biochemical genetics, (2017); 55(5-6): 410-420.
- Bukhari I, Mujtaba G, Naz S. Contribution of GJB2 mutations to hearing loss in the Hazara Division of Pakistan. Biochemical genetics, (2013); 51(7-8): 524-529.
- Santos R, Wajid M, Pham T, Hussan J, Ali G, et al. Low prevalence of Connexin 26 (GJB2) variants in Pakistani families with autosomal recessive non‐syndromic hearing impairment. Clinical genetics, (2005); 67(1): 61-68.
- Anjum S, Azhar A, Tariq M, Baig SM, Bolz HJ, et al. GJB2 Gene Mutations Causing Hearing Loss in Consanguineous Pakistani Families. Pakistan Journal of Life & Social Sciences, (2014); 12(3).
- Salman M, Bashir R, Imtiaz A, Maqsood A, Mujtaba G, et al. Mutations of GJB2 encoding connexin 26 contribute to non-syndromic moderate and severe hearing loss in Pakistan. European Archives of Oto-Rhino-Laryngology, (2015); 272(8): 2071-2075.
- Doll J, Vona B, Schnapp L, Rüschendorf F, Khan I, et al. Genetic spectrum of syndromic and non-syndromic hearing loss in Pakistani families. Genes, (2020); 11(11): 1329.
- Nisar S, Tariq M, Adeel A, Gogate M, Hussain A. Cognitively inspired feature extraction and speech recognition for automated hearing loss testing. Cognitive Computation, (2019); 11(4): 489-502.
- Tariq H, Zaigham K, Kousar S, Azhar A. Genetic contribution of GJB2 gene to hearing impairment in Pakistan. Advancements in Life Sciences, (2019); 7(1): 38-43.
- Zafar S, Shahzad M, Ishaq R, Yousaf A, Shaikh RS, et al. Novel Mutations in CLPP, LARS2, CDH23, and COL4A5 Identified in Familial Cases of Prelingual Hearing Loss. Genes, (2020); 11(9): 978.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0

