

Full Length Research Article
Impact of Sickle Cell Anemia on children growth and clinical parameters in Al-Ahsa region of Saudi Arabia
Nawaf Alanazi1’2, Shahad Alabdullatif2, Maryam Albahrani2, Malak Aljamaan2, Fatimah Alsayegh2, Aysha Bhalli2, Khaled Aljarrah3,4, Zafar Iqbal1,2*
Adv. life sci., vol. 8, no. 2, pp. 179-183, February 2021
*- Corresponding Author: Zafar Iqbal (Email: iqbalz@ksau-hs.edu.sa)
Authors' Affiliations
2. Clinical Laboratory Sciences Program, College of Applied Medical Sciences, King Saud Bin Abdulaziz University for Health Sciences / KAIMRC/ SSBMT, King Abdulaziz Medical City, National Guard Health Affairs, Al-Ahsa – Saudi Arabia
3. Department of Physics, Jordan University of Science and Technology, Irbid – Jordan
4. Department of Basic Sciences, College of Applied Medical Sciences, King Saud Bin Abdulaziz University for Health Sciences & KAIMRC, King Abdulaziz Medical City, National Guard Health Affairs, Al-Ahsa - Saudi Arabia
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Sickle cell disease (SCD) is an autosomal recessive disease caused by a single gene mutation, leading to sickle-shaped red blood cells, causing many clinical complications. Resulting complications may affect the growth of the SCD patients that is a strong measure of severity of disease and helps in disease management strategies in any area. Eastern province of Saudi Arabia has one of the highest SCD incidences. Nevertheless, no studies have been previously carried out of about clinical outcome of SCD in Al-Ahsa area of eastern province. Therefore, this study was conducted to find out the impact of SCD children at king Abdulaziz Hospital Al-Ahsa.
Methods: All pediatric SCD patients were included in the study. Patient data was taken from hospital information system and analyzed using SPSS version 27.
Results: A total of 53 patients were studied. The male to female ratio was 1.4:1 and mean age was 3.3 years (range: 1-9). Eighteen (34%) did not present with sickle cell crisis possibly due to ameliorating effects of high HbF and G6PD deficiency. Although growth parameters of SCD patients were not statistically different from international standards, there was significant difference between weight of SCD patients in recurrent sickle cell crisis group and non-crisis sickle cell (NC-SC) group at diagnosis and after clinical interventions (p= 0.04 and 0.03, respectively) that included hydroxyurea. The corrected reticulocyte (at diagnosis and after clinical intervention) and WBC counts were statistically significant between hydroxyurea and non-hydroxyurea groups (p-value < 0.05).
Conclusions: Overall, one-third of SCD patients in Al-Ahsa region have mild disease and hydroxyurea can minimize the SCD severity through lowering corrected reticulocyte and WBC counts. Exact mechanisms of mild SCD and hydroxyurea in minimizing disease severity are needed to be elucidated.
Keywords: Salinity; Livelihood; Productivity; Farming experience; Farm size
Introduction![]()
Sickle cell anemia is an inherited disease with the production of sickled red blood cells and hemoglobin S instead of hemoglobin A [1]. Consequently, hemoglobin’s ability of transferring oxygen all over the body decreases. Moreover, normal red blood cells’ life span is 120 days, but in sickle cell anemia, red blood cells last for 10-20 days [2,3] The diagnosis of sickle cell anemia is performed by a hemoglobin electrophoresis to check for hemoglobin S and this blood test is part of the routine newborn screening in Saudi Arabia.
Sickle cell anemia patients experience many symptoms that vary from person to person which include fatigue, episodes of pain, painful swelling of limbs, frequent infections, delayed growth, and vision problems [4]. Sickle cell anemia affects a lot of body functions and causes some serious complications. The most common complications among sickle cell anemia patients are the retention of blood in limbs due to blood vessels obstruction, and this is known as hand-foot syndrome [5]. The sickled cells stick to wall of blood vessels and block the blood flow to the different body organs. When blood vessels are blocked, oxygen and nutrients will not be able to reach organs, and that will lead to organ dysfunction and death. A cure for sickle cell anemia is unavailable, so medication is usually taken to help with complications. For instance, hydroxyurea is administered to prevent pain [6]. In case the patient is having a pain episode, medication is given based on the severity of the crisis like aspirin for mild to moderate pain and opioid (morphine) for severe pain. Blood transfusions are effective to compensate the function of the sickled cells [6].
Sickle cell disease can severely affect growth of the children, in addition to other clinical manifestations [2, 4]. There are different reports about impact of sickle cell disease on children growth. A study carried in India in 2004 reported that children with sickle cell anemia had lower weight and height [7, 8]. In another study conducted in US on children and adolescents aged 6 to 18 years, no significant changes in height or weight of SCD children were noted as compared to healthy population [9]. A study conducted in Philadelphia showed that children with sickle cell anemia had low bone mineral density maturation [10]. Studies have shown that sickle cell disease can have varying effects on growth and other health parameters in different ethnic groups that may be due to different SCD phenotypes in different geographic regions of the world [11]. Moreover, various clinical complications resulting due to SCD may affect the growth of the SCD patients that is a strong measure of severity of disease and helps in disease management strategies in any area.
The incidence of SCD and its phenotypes vary in different parts of Saudi Arabia [8, 12]. The prevalence of SCD trait varies from 2-27% in different parts of Saudi Arabia while disease has a severe and a mild phenotype [8, 12]. Although many studies have been conducted in Saudi Arabia about sickle cell disease, no study has been carried out in Al-Ahsa region about impact of sickle cell anemia on growth parameters (height, weight) of sickle cell patients. Therefore, objective of this study is to find the effect of sickle cell anemia on height, weight and of children at King Abdulaziz Hospital in Al-Ahsa region.
Methods![]()
This study was carried out retrospectively, and it was approved from institutional research committee and ethical review board. Clinically diagnosed SCD patients in children age groups (1-15 years) were included in the study. Adult SCD patients as well as patients with hemoglobinopathies other than SCD were excluded. Patient data was retrieved from the medical records at Division of Hematology/Oncology, Department of Pediatrics, King Abdulaziz Hospital, Al-Ahsa, Eastern region, Saudi Arabia. The data was analyzed using SPSS version 27. The growth parameters were compared between these different patient groups. The paired t-test was used to compare groups. P-values of 0.05 were considered to be significant.
Results![]()
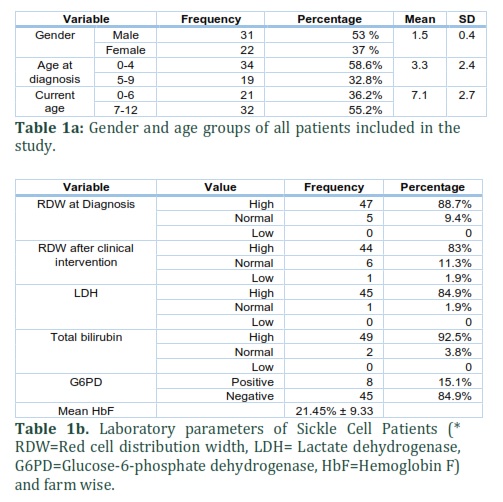
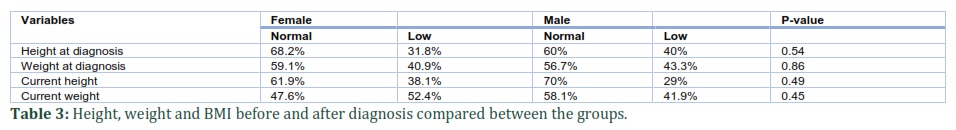
This study included 53 children diagnosed with sickle cell disease (SCD) in King Abdulaziz Hospital Al-Ahsa from 2013-2018. The age group for the selected sample size is from 0-12 years. The numbers of males were 31 (53%) and females were 22 (37%). The male to female ratio was 1.4:1. Moreover, most of the children diagnosed with SCD belonged to age group 0-4 (58.6%) and lowest to age group 5-9 years (32.8%). (Table 1a). Glucose-6-phosphate dehydrogenase deficiency (G6PD) was found in 5 (9.4%) patients while mean HbF was 21.45% ± 9.33 (Table 1b). Out of 53 patients, 18 (34%) did not present with sickle cell crisis (non-crisis sickle cell group). All patients without sickle cell crisis had either high HbF level or G6PD deficiency. Therefore, high HbF levels and G6PD deficiency may have ameliorating effects on sickle cell patients.
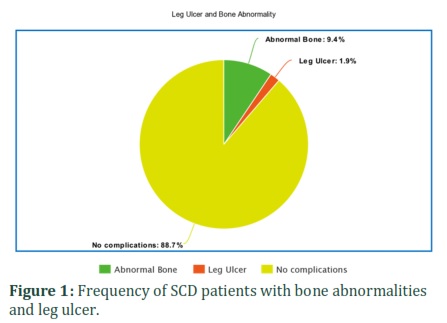
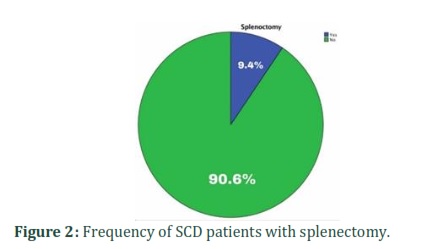
Patients included in this study had a variety of moderate to severe complications caused by the disease. One patient (1.8%) was observed with leg ulcer. Bone X-ray images showed that 8.6% of patient bad bone abnormalities. Splenectomy is a therapeutic procedure that can be done in some severe sickle cell cases. Overall, 8.6% of patients with sickle cell disease had their spleen surgically removed (Figure 1 & Figure 2).
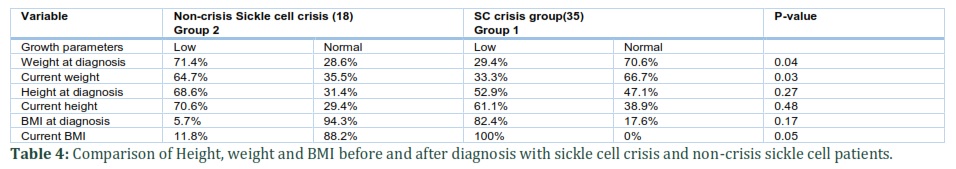
Overall, most of the male and female SCD patients had normal height and weight when compared with the international height and weight standards for healthy children (Table 2 and Table 3).
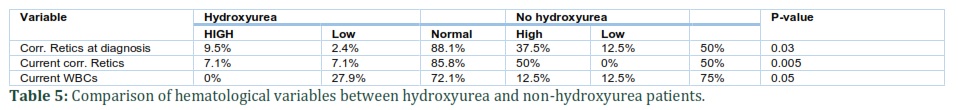
Furthermore, the patients were divided into two groups, the patients suffering from recurrent sickle cell crisis (RC-SC) group (35/53=66%) and patients with non-crisis sickle (NC-SC) cell group (18/53=34%). Chi-square test results showed that there was significant difference in weight of the patients in two groups at diagnosis and after clinical intervention (p-values: 0.04 and 0.03, respectively) (Table 4). Moreover, there was no statistically significant difference in height of both patient groups and body-mass index (BMI) at diagnosis (Table 4). Nevertheless, significant difference in BMI of both groups was noted after clinical intervention (p=0.05).
Crisis and non-crisis sickle cell patients
Not all patients agree to hydroxyurea treatment. Treatment with hydroxyurea had on impact on sickle cell crisis management. Among group I patients (RC-SC) patients, 71.1% were taking hydroxyurea while 37.5% group 2 patients (NC-SC) provided consent for hydroxyurea urea. (P-value: 0.06). It necessitates needs to counsel all SCD patients and their families to take hydroxyurea.
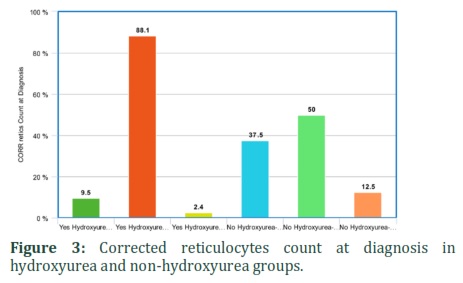
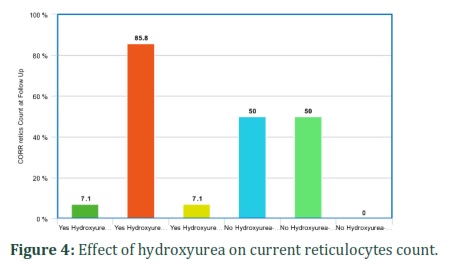
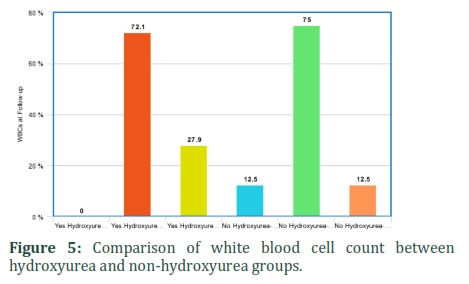
It was also found that hydroxyurea significantly affected corrected reticulocytes and WBC count in SCD patients. Corrected reticulocytes count in both at diagnosis and current was higher in non-hydroxyurea group (50%) as compared to hydroxyurea group (7.1%). WBCs count was low in 27.9% of patients taking hydroxyurea as compared to 12.5% of non-hydroxyurea group patients. The hematological parameters like corrected reticulocyte count (diagnosis and current) and WBC were statistically significant between hydroxyurea and non-hydroxyurea groups (p-value: 0.03, 0.005 and 0.05) (Table 5) (Figures 3-5). It shows that hydroxyurea lowers reticulocytes and WBC count in sickle cell patients that can help in reducing severity of disease. Exact mechanisms involved in these beneficial effects of hydroxyurea for sickle cell patients through lowering down reticulocyte and WBC counts are needed to be investigated.
Among all growth parameters the current weight, height and BMI were lower in SCD patients not taking hydroxyurea as compared to hydroxyurea group. However, the p-value is more than 0.05 which is statistically not significant (Table 6). Studies with higher number of patients may explain the exact correlation of these growth parameters with hydroxyurea treatment.
Overall, one third of SCD patients in Al-Ahsa region have mild disease and hydroxyurea can minimize the SCD severity through lowering corrected reticulocyte and WBC counts. Exact mechanisms of mild SCD and hydroxyurea in minimizing disease severity are needed to be elucidated.
Figures & Tables


Discussion![]()
The aim of the study was to investigate the impact of sickle cell disease on children from Al-Ahsa region, Eastern Province, Saudi Arabia. Our study shows that 34% patients had no sickle cell crisis or other severe clinical manifestations. This is in accordance with previous studies reporting milder SCD in Eastern Province of Saudi Arabia and other regions [8, 12]. All of the SCD patients with milder disease had either high HbF (fetal hemoglobin) level or G6PD deficiency. Several other studies have reported protective effects of high HbF levels and G6PD deficiency in SCD [13, 14]. Level of HgF is controlled by multiple genetic loci likeHBB cluster, BCL11A, and HMIP-2 (HBS1L-MYB) and expression of these genes is under tight genetic control as manifested by involvement of many gene repressors, enhancers and other factors. Silencing of repressors of HgF-inducing genes, for example, BCL11A repressor, is being investigated as innovative therapeutic strategy to develop novel medications for inducing high protective levels of HgF in SCD with initial promising results [15]. Several HgF-inducing drugs like hydroxyurea are already in clinical use [13, 15].
There are ethnic and phenotypic variations in impact of SCD on growth parameters of patients in different parts of the world [9-11]. In our studies, overall male and female SCD patients had normal height and weight in comparison to international height and weight standards for healthy children. This is in accordance with a study published from USA, that reported no significant changes in height or weight of SCD on children and adolescents aged 6 to 18 years as compared to healthy population [9]. Nevertheless, weight was significantly lower in sickle cell crisis group as compared to non-crisis SCD group at diagnosis (p=0.03) as well as after clinical interventions (0.04) in our studies. It could be Furthermore, non-crisis SCD group had more improvement in weight, height and BMI as compared to sickle-cell crisis group but it was not significant. It is understood as vaso-occlusive crisis is the major reason of major complications in SCD [16].
Hydroxyurea is the drug of choice for SCD patients, specifically for patients with vaso-occlusive crisis, but not all patients provided consent to take this medication. It is utilized as inducer of HgF expression that has protective effects for SCD patients [13, 14]. Our studies showed that SCD patients receiving hydroxyurea treatment had more improvement in body-mass index as compared to patients not receiving hydroxyurea. Similar results have been reported from investigators [17]. Our studies further showed decreased in WBC and corrected reticulocytes count in SCD patients receiving hydroxyurea, which is in accordance with other reports [17,18]. Although there is correlation between SCD, hydroxyurea treatment, lowering of WBC/reticulocytes and clinical improvement of SCD complications, exact mechanism of action of hydroxyurea still remains to be elusive [17-19]. Further studies by employing state-of-the-art multi-omics technologies are required to find out factors associated with milder SCD in Al-Ahsa region of Saudi Arabia and to unravel cellular molecular mechanism of action in of hydroxyurea in SCD patients.
Our study shows 18 (34%) did not present with sickle cell crisis possibly due to ameliorating effects high HbF levels and G6PD deficiency. Although growth parameters of SCD patients were not statistically different from international standards, there was significant difference between weight of SCD patients in recurrent sickle cell crisis group and non-crisis sickle (NC-SC) cell group at diagnosis and after clinical intervention (p-values: 0.04 and 0.03, respectively) that included hydroxyurea in eligible patients. The corrected reticulocyte (at diagnosis and after clinical intervention) and WBC counts were statistically significant between hydroxyurea and non-hydroxyurea groups (p-value < 0.05). Overall, one third of SCD patients in Al-Ahsa region have mild disease and hydroxyurea can minimize the SCD severity through lowering corrected reticulocyte and WBC counts. Exact mechanisms of mild SCD and hydroxyurea in minimizing disease severity are needed to be elucidated.
Author Contributions
All authors have contributed to the article per international requirements of the authorship. The manuscript has been read and approved by all the authors, and the requirements for authorship have been met, and that each author believes that the manuscript represents original and honest work.
The authors declare that they have no competing interests.
References![]()
- Pecker LH, Lanzkron S. Sickle Cell Disease. Annals of Internal Medicine, (2021); 174(1): ITC1-ITC16.
- Piccin A, Murphy C, Eakins E, Rondinelli MB, Daves M, Vecchiato C, et al. Insight into the complex pathophysiology of sickle cell anaemia and possible treatment. European Journal of Haematology, (2019); 102(4):319-330.
- Faes C, Ilich A, Sotiaux A, Sparkenbaugh EM, Henderson MW, Buczek L, et al. Red blood cells modulate structure and dynamics of venous clot formation in sickle cell disease. Blood, (2019) ;133(23):2529-2541.
- Kjellander C, Sennström MK, Stiller V, Ågren A. Sicklecellanemi ger skiftande symtombild och hög morbiditet–Allvarlig prognos vid världens vanligaste genetiska sjukdom [Sickle cell anemia causes varied symptoms and high morbidity. Serious prognosis in the most common genetic disease in the world]. Lakartidningen. (2015);112:DCPM.
- Williams TN, Thein SL. Sickle Cell Anemia and Its Phenotypes. Annual Review of Genomics and Human Genetics, (2018); 19: 113-147.
- Kapoor S, Little JA, Pecker LH. Advances in the Treatment of Sickle Cell Disease. Mayo Clinic Proceedings (2018); 93(12): 1810-1824.
- WHO Multicentre Growth Reference Study Group. WHO Child Growth Standards based on length/height, weight and age. Acta Paediatrica, (2006);450:76-85.
- Jastaniah W. Epidemiology of sickle cell disease in Saudi Arabia. Annals of Saudi Medicine, (2011);31(3):289-93.
- Mitchell MJ, Carpenter GJ, Crosby LE, Bishop CT, Hines J, Noll J. Growth status in children and adolescents with sickle cell disease. Pediatric Hematology Oncology, (2009); 26(4): 202-15.
- Adesina OO, Gurney JG, Kang G, Villavicencio M, Hodges JR, Chemaitilly W, et al. Height-corrected low bone density associates with severe outcomes in sickle cell disease: SCCRIP cohort study results. Blood Advances, (2019) 14; 3(9): 1476-1488.
- Adegoke SA, Figueiredo MS, Adekile AD, Braga JAP. Comparative study of the growth and nutritional status of Brazilian and Nigerian school-aged children with sickle cell disease. International Health, (2017); 9(6): 327-334.
- Al-Suliman A, Elsarraf NA, Baqishi M, Homrany H, Bousbiah J, Farouk E. Patterns of mortality in adult sickle cell disease in the Al-Hasa region of Saudi Arabia. Annals of Saudi Medicine, (2006); 26(6):487-8.
- Sales RR, Belisário AR, Faria G, Mendes F, Luizon MR, Viana MB. Functional polymorphisms of BCL11A and HBS1L-MYB genes affect both fetal hemoglobin level and clinical outcomes in a cohort of children with sickle cell anemia. Annals of Hematology, (2020); 99(7):1453-1463.
- Belisário AR, Sales RR, Toledo NE, Velloso-Rodrigues C, Silva CM, Viana MB. Glucose-6-Phosphate Dehydrogenase Deficiency in Brazilian Children With Sickle Cell Anemia is not Associated With Clinical Ischemic Stroke or High-Risk Transcranial Doppler. Pediatric Blood & Cancer, (2016);63(6):1046-1049.
- Esrick EB, Lehmann LE, Biffi A, Achebe M, Brendel C, Ciuculescu MF, et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. The New England Journal of Medicine, (2021); 384(3): 205-215.
- Shah N, Bhor M, Xie L, Paulose J, Yuce H. Sickle cell disease complications: Prevalence and resource utilization. PLoS One, (2019); 14(7): e0214355.
- Hankins JS, Aygun B, Nottage K, Thornburg C, Smeltzer MP, Ware RE, et al. From infancy to adolescence: fifteen years of continuous treatment with hydroxyurea in sickle cell anemia. Medicine (Baltimore), (2014); 93(28): e215.
- Rana S, Houston PE, Wang WC, Iyer RV, Goldsmith J, Casella JF, et al. Hydroxyurea and growth in young children with sickle cell disease. Pediatrics, (2014); 134(3): 465-72.
- Pule GD, Mowla S, Novitzky N, Wiysonge CS, Wonkam A. A systematic review of known mechanisms of hydroxyurea-induced fetal hemoglobin for treatment of sickle cell disease. Expert Review of Hematology, 2015; 8(5): 669-79.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0


