

Review Article
Next Generation Sequencing as Rapid Diagnosis of Multidrug Resistance Tuberculosis
Anse Diana Valentiene Messah1, Jeanne Adiwinata Pawitan2,3,4*
Adv. life sci., vol. 8, no. 1, pp. 30-37, November 2020
*- Corresponding Authors: Jeanne Adiwinata Pawitan (Email: jeanneadiwip@gmail.com)
Authors' Affiliations
2. Department of Histology, Faculty of Medicine, Universitas Indonesia – Indonesia
3. Stem Cell Medical Technology Integrated Service Unit, Dr. Cipto Mangunkusumo General Hospital/Faculty of Medicine Universitas Indonesia, – Indonesia
4. Stem Cell and Tissue Engineering Research Center, Indonesia Medical Education and Research Institute (IMERI), Faculty of Medicine Universitas Indonesia – Indonesia
Abstract![]()
Introduction
Methods
Discussion
Conclusion
References
Abstract
Multi-drug-resistant tuberculosis (MDR-TB) is a threat to global health. In 2018, TB related death was estimated to be more than 1.5 million cases worldwide. Conventional diagnostic method, which requires a long time to get a result, causes delays in new cases discoveries that lead to delayed therapy. Further, delayed and inadequate therapy causes an increase in the level of resistance to anti-TB drugs that may lead to death. Therefore, diagnostic tools, which can detect quickly and accurately, are highly needed. Early and timely detection is crucial for globally effective TB control, but this is not popular in developing countries, especially in Asia. Therefore, the objective of this review is to provide current information on the use of NGS as a rapid diagnostic tool for MDR-TB, especially in Asian populations, and to highlight the various MDR genes.
Keywords: Next Generation sequencing; Multi-Drug Resistance; Tuberculosis; Mycobacterium tuberculosis; Rapid diagnosis
Introduction![]()
In 2018, tuberculosis (TB) new cases and TB related death were estimated to be 10 million and 1.5 million cases respectively. There was a 2% annual decline in global TB incidence between 2017 and 2018, but this decline is far from enough to achieve the world’s pledge to end TB epidemic by 2030 [1]. Though global TB incidence and mortality rates have declined recently, efforts to control TB were hampered by increased multi-drug resistant TB (MDR-TB), which were resistant to at least two first-line drugs (isoniazid, rifampicin, and ethambutol), especially in Asian countries [2, 3].
World Health Organization (WHO) reported that there were thirty countries with high TB burden, and among those countries twelve were located in Asia, i.e. Vietnam, Thailand, Philippines, Papua New Guinea, Pakistan, Myanmar, Indonesia, India, Korea, China, Cambodia and Bangladesh. Collectively, the Philippines (6%), Indonesia (8%), China (9%), and India (27%) contributed to half of all TB cases, and approximately 44% was contributed by the Southeast Asia region [2, 4].
Tuberculosis cases are increasing every year due to to the resistance developed by Mycobacterium tuberculosis against anti-tuberculosis drugs, especially in Asia. Conventional diagnostic method, which needs culturing of the bacteria, requires a long time to get a result, and causes delays in new cases discoveries that lead to delayed therapy. Further, delayed and inadequate therapy causes an increase in the level of resistance to anti-TB drugs that may lead to death. Therefore, diagnostic tools, which can detect quickly and accurately, are highly needed.
Successful diagnosis and treatment of MDR-TB depends on drug susceptibility testing (DST). Conventionally, the diagnosis of drug resistance to Mycobacterium tuberculosis (MTB) is highly dependent on culture and DST in liquid or solid media. Those phenotypic results can be obtained after weeks to months of incubation. In addition, phenotypic testing are often inaccurate, especially for drugs such as pyrazinamide [5].
Identification and characterization using molecular test on genetic basis to detect pathogenic mutations in MDR-TB DNA samples have been used for diagnosis, determining prognosis, and prospective therapy. The latest development of next generation sequencing (NGS) has revolutionized the molecular test for MDR-TB diagnosis, especially in developed countries. NGS presents options for detecting the characteristics of mutation in MDR-TB [6, 7] that is not readily available in most Asian countries.
Therefore, this review aimed to provide current information on the use of NGS as a rapid diagnostic tool of MDR-TB especially in Asian population, and to highlight the various MDR genes. For this purpose, we discussed MDR-TB, molecular mechanism of drug resistance in TB, MDR-TB diagnostic problems and solutions, NGS for whole-genome sequencing to diagnose MDR-TB, various NGS platforms, NGS methods, and application of NGS to detect variants of drug-resistant MTB especially in the Asian population.
Methods![]()
Literature Search strategy and selection criteria
Literature search was done in PubMed/Medline and Google Scholar on 20th of May 2020, and the keywords used were combination of: “multidrug resistance”, “tuberculosis”, “diagnosis”, “Asian population” and “next generation sequencing”. All articles that provided information on MDR-TB, molecular mechanism of drug resistance in tuberculosis, MDR-TB diagnostic problems and solutions, various means to diagnose MDR-TB including NGS, various NGS platforms, NGS methods, and application of NGS to detect variants of drug-resistant MTB were included. Where possible, articles were restricted to recent articles that were published in last 10 years, except the content was very important. In addition, existing suitable article in authors’ files were added. Finally, 47 articles were selected to write this review.
Discussion![]()
Multidrug resistant TB
Primary TB is a disease due to MTB in a patient who has never taken anti-TB medication or has taken for less than a month [8]. Endogenous reactivation occurs when there is a reactivation of a previous infection that was contained by the host immune response [9]. Drug-resistant TB is resistant to a usually used anti-TB. The way of drug-resistant TB transmission is the same as drug-susceptible TB [10]. MTB causes TB infection and is contagious in the form of MTB containing airborne particles, which are called droplet nuclei, of 1– 5 microns in diameter. MTB containing droplet nuclei are generated when a person with pulmonary or laryngeal TB sneezes, coughs, sings or shouts. TB transmission occurs when MTB containing droplet nuclei are inhaled by a person through the mouth or nose, and finally reach the alveoli of the lungs [11].
Drug-resistant TB can develop in two different ways that results in primary and secondary resistance. Primary resistance occurs due to initial infection with resistant organisms. Secondary resistance, or acquired resistance occurs during TB therapy, either due to an inadequate regimen, or the prescribed regimen is not taken appropriately. Conditions in which a person has an increased risk to contract drug-resistant TB are: exposure to a drug-resistant TB patient, or a TB patient who has got prior TB treatment but the treatment failed or relapse, and whose susceptibility test results are not known, or to a TB patient from an area with a high prevalence of drug resistance, or travel to one of these areas, or exposure to a person with continuous positive smear and cultures after two months of combination TB therapy [10, 12].
Molecular mechanism of drug resistance in tuberculosis
There are two mechanisms of drug resistance in TB, intrinsic and acquired drug resistance. Intrinsic drug resistance of MTB is attributed to the structure of MTB wall, or the ability of MTB to produce a certain enzyme. Intrinsic resistant MTB has mycolic acid containing cell wall, which causes low permeability to anti-tuberculosis drugs that is an important factor for natural antibiotic resistance. In addition, MTB can produce a β-lactamase, which is an enzyme that damages antibiotics with beta-lactam structure, where the β-lactamase is coded by blaS and blaC [12-14]. The acquired drug resistance is due to an inappropriate drug regimen or suboptimal TB treatment, which triggers spontaneous genetic mutations in chromosome genes, and causes resistance. The rate of mutation for most anti-TB drugs for one TB drug per cell division is around 10-9 and will be 10-18 for two kinds of TB drugs; thus this fact is the main reason why anti-TB drugs are given in combination [15].
Mutations that cause isoniazid resistance may occur in various genes, namely katG, ahpC, inhA, kasA, ndh and fabG. KatG encodes catalase-peroxidase, which is a bifunctional enzyme, whose peroxidase has a broad spectrum activity that oxidize various electron donors, including NADP(H); AhpC encodes Alkyl hydroperoxide reductase C that functions in protecting cells against oxidative stress by peroxide detoxification, which is the main scavenger for endogenous hydrogen peroxides; InhA encodes NADH Enoyl-[acyl-carrier-protein (ACP)] reductase, which is essential for mycolic acid synthesis; kasA encodes 3-oxoacyl-[ACP] synthase 1, which catalyzes fatty acid synthesis condensation reaction due to an addition of two carbons from malonyl-ACP to an acyl acceptor; Ndh encodes NADH dehydrogenase, which transfer electron from NADH in the respiratory chain to ubiquinone, where this redox reaction does not coincide with proton translocation. Two main molecular mechanisms that cause isoniazid resistance are mutations in the katG and inhA, which most often occurs in the promoter region of the katG, and inha genes. Activated isoniazid interferes with the synthesis of mycolic acid through inhibition of NADH enoyl-ACP reductase, which is encoded by inhA, and causes the MTB cell wall to become more permeable to anti-TB drugs [15-19]. Therefore, mutations in promoter region of fabG-inhA operon, which causes overexpression of inhA or decrease in inhA affinity to activated isoniazid, contribute to low-level resistance. Mutations in fabG-inhA operon are less frequent than mutations in the katG [20].
Rifampicin binds MTB RNA polymerase b-subunit, which is encoded by rpoB gene, and inhibits messenger RNA elongation. Therefore, mutations in the rpoB gene cause rifampicin resistance, which are found in most rifampicin resistant MTB clinical isolates [15-19]. The main mechanism in MTB pyrazinamide resistance is a mutation in pncA gene that encodes pyrazinamidase. Pyrazinamidase is an enzyme, which converts pyrazinamide into pyrazinoic acid that is active in inhibiting MTB. Therefore, mutations in pncA abolish the conversion ability and cause pyrazinamide resistance, which is revealed by a strong correlation between pncA mutations and MTB resistance to pyrazinamide. The mutations mostly occur at an open reading frame of 561 bp, or in an area of 82 bp that is suspected as a promoter [21].
Mutations in MTB for ethambutol resistance are less studied compared to those of isoniazid, rifampicin, and pyrazinamide. Mutations in embB i.e. in codons 406, 354, 319 and 306 were associated with ethambutol resistance [22].
Second-line drugs for the treatment of TB are fluoroquinolones including ofloxacin, levofloxacin, and moxifloxacin. The target of fluoroquinolone in MTB is type II DNA gyrase (topoisomerase). DNA gyrase, which is a tetramer, consists of subunits A and B that is encoded by gyrA and gyrB genes. Both genes are often found to be associated with fluoroquinolone resistance [10, 23].
Another second line drugs for MDR-TB are aminoglycosides such as kanamycin, amikacin, and capreomycin. Kanamycin and amikacin bind to 16S rRNA of small 30S ribosome subunits, thus cause protein synthesis inhibition. The 16S rRNA is encoded by rrs. The most common mutations occurred in A1401G rrs that were identified in 30-90% of amikacin and kanamycin resistant MTB strains. Capreomycin inhibits phenylalanine synthesis in MTB ribosome, thus causes translation inhibition. Phenylalanine ribosomal synthesis is also encoded by rrs. Therefore, mutations in the region of rrs gene cause capreomycin resistance. Besides that, capreomycin resistance can also occur when there is a mutation in the promoter of eis gene, which encode aminoglycoside acetyltransferase, in the form of low-level point mutation [10, 24].
MDR-TB diagnostic problems and solutions
Until now, MDR-TB is one of the global problems in the world, especially in low-income countries like Asia. The highest number of TB cases, which was in India followed by Indonesia, was reported by WHO in 2017 [2]. Moreover, Indonesia has the highest levels MDR-TB and HIV in the world. This number continues to increase every year due to inadequate drug therapy and high resistance to anti-TB drugs. Though rapid molecular diagnostic test kits are available, it cannot be used to explain the variation and mechanism of resistance to anti-TB drugs. Most diagnostic tests only examine one or a certain combination of genes, which bear the most common mutation or hotspot genome area, thus a sample need several tests to get a complete drug resistance pattern that is needed to choose appropriate drug combination for the patient. Ideally, a diagnostic tool should provide complete information of whole MTB genome [7, 8, 19]. The rapid development in molecular biology may provide information of whole MTB gene sequence [7, 8].
Information of whole MTB genome may increase knowledge on the mechanism of resistance to major anti-TB drugs, as it may show the relation of a specific gene mutation with drug resistance. Therefore, the molecular mechanism of a specific drug resistance is known, and can be used to improve the currently available techniques for rapid detection of MDR-TB [23, 25].
NGS for whole genome sequencing to diagnose MDR TB infection
Currently, next-generation sequencing (NGS) is widely used for drug resistance rapid diagnosis, so that prospective treatment and prognosis of patients with MDR-TB can be determined. Moreover, NGS can be used to sequence the whole-genome of MTB. However, there are some challenges in using NGS i.e. sample preparation is elaborated, so the methodology needs to be simplified and optimized for direct sputum samples; in some developing countries, there is lack of capacity, and high cost may hamper its use in global surveillance [3].
NGS provides options to detect the whole characteristics of MDR-TB. Unlike other molecular tests for MDR-TB, which relies on indirect MTB identification by a series of limited resistance mutations through hybridization of a probe to a specific genetic sequence, the NGS may provide detailed whole genome information. In NGS, each genome parts are sequenced several times, so this assessment provides accurate data and insight into the presence of genetic polymorphisms [26]. Therefore, NGS can confirm and assess genome level mutations that cannot be detected by other molecular tests. Moreover, NGS is flexible and can be programmed for various applications, such as for assessment of genetic information of co-existing micro organisms other than MTB that is present in a clinical sample [10].
Despite the challenges, the benefits of NGS remain more compared to other methods, when they are used for diagnosis, to determine appropriate treatment, and epidemiology. The benefits of NGS include time for faster diagnosis of MTB (from weeks in other methods to hours in NGS), the amount of information provided (information about strains, whether single or multiple infection; distinguishing recurrence from re-infection; drug resistance profile; and chain of transmission) and comfort (potential to combine diagnosis, drug resistance, and epidemiological analysis) that can be collected in real-time. In addition, NGS can be done on direct clinical specimens or cultured MTB isolates [27].
Apart from the benefits over other molecular methods, the use of NGS is hindered in low and middle-income countries, as NGS needs high cost, trained operator, special means for data analysis/storage, and plug-and-play device to directly sort information from primary clinical samples, which are not readily available in those countries [10, 27].
Both phenotype and genotype tests have been used to detect MTB drug resistance to various types of drugs. The phenotype test is obtained by culture, which requires a long time from several weeks to months, while genotyping may shorten the time, so this method, especially those using NGS is highly needed [28].
Whole Genome Sequencing (WGS) is a method to determine the nucleotide sequence of certain genomes, which are the genetic material of whole cells or microbes.28 This method might obtain genetic and epidemiological data, as well as indications of cross contamination risk, with high sensitivity [29, 30]. The WGS provides more precise and accurate information and improves understanding of MTB pathogenesis, immunology, and transmission [31].
WGS requires a genome as a reference to show various genetic mutations/sample variations. The genome, which is the most used as a reference, is MTB H37Rv genome with worldwide distribution and wide usage. The H37Rv genome is one of the best for curating MTB, due to transcriptomic and proteomic data sources that are available in the world. In molecular epidemiology and phylogenetic studies, mutations are associated with anti-tuberculosis drug resistance in MTB [23].
Various NGS platforms
NGS, which is a "high throughput, massive and parallel" sequencing method, can be used to assess the sequence of a whole genom where all genes are ordered along the whole target genom in a single biochemical reaction. This technique sort multiple DNA fragments in parallel, assemble and map them according to the reference genome using bioinformatic analysis [10, 32].
To obtain the maximum potential of NGS in a clinical setting to improve TB diagnosis and management, the NGS workflow and platform should be understood, so that optimal integration of NGS into existing laboratory workflows can be achieved [6, 33]. There are two main NGS platforms, i.e. that works by producing relatively short DNA sequences (<1000 base pairs), or longer DNA sequences (long reads of > 5,000 base pairs in average). The speed to get detailed information from NGS holds the potential to increase the accuracy of MDR-TB diagnosis, speed up TB treatment, as well as better understand TB epidemiology. NGS provides information in the form of thousands of millions of varying length reading to be assembled into longer sequences or even the whole genome, using bioinformatics. In addition, NGS platforms might use different sequencing chemistry (Table 1) [34, 35].

All NGS platforms use similar basic workflow to get sequence readings from clinical samples, which consist of: DNA extraction, purification, target specific enrichment, and data analysis. DNA extraction from clinical samples (sputum/sediments) or cultured isolates should have sufficient yield, integrity, and purity. High quality DNA is crucial in NGS, especially WGS, thus efficient DNA extraction and purification methods are highly important. The DNA extraction method should be able to break the MTB cell wall to release the DNA to be purified. After purification, the next step is target specific enrichment. The resulting DNA fragments are then sorted in parallel, followed by data analysis, where the fragments are mapped according to the reference genome using bioinformatics special software [10].
Whole genome sequencing can be used as a tool to determine sequence variations in epidemiology and to determine the source of infection and transmission pattern between various patients. WGS uses NGS technology that can generate millions of brief readings from the entire genome, which is then analyzed by a special software [32].
Specific target enrichment
Next generation sequencing needs to do specific target enrichment that aims to eliminate genomic DNA regions that are not of interest in a certain experiment. By targeting specific areas, a greater sequence information is obtained for the areas of interest. In addition, time, resources and cost can be reduced, so that more individual samples can be analyzed. The specific target enrichment strategy can be carried out by various approaches to capture specific sequences of interest, i.e: hybrid capture, selective circularization, and PCR amplification [36].
Hybrid Capture is conducted using nucleic acid sequences, which are derived from input samples, to be specifically hybridize to pre-prepared DNA fragments that are complementary to the target area of interest, so that the target area of interest can be captured, isolated, and enriched, either on a solid support or in solution [36].
Selective circularization is conducted using molecular inversion probes (MIP), or gap-fill padlock or selector probes in the form of single strand (ss) DNA circles, which contain the target areas to be enriched. The ss DNA circles, which are formed by gap filling and ligation in such a way, are used for selective amplification of the target area of interest [36].
PCR Amplification is conducted using polymerase chain reaction (PCR) method, which is directed to the target areas of interest, by carrying out many remote PCRs in parallel, in limited quantities. The PCR method is very multiplex that amplifies a very large number of short fragments [36].
Next generation sequencing methods
There are several NGS methods that need to be known and are often used for diagnostic tools. They are WGS, targeted and shotgun NGS. The first method is WGS.
WGS is a method to analyse the entire sequence of a cell whole genome at a single time to provide the most comprehensive information of the genome. Therefore, WGS provides the most comprehensive data on a certain organism, due to its ability to show the profile of an entire genome; thus WGS enables scientists to find genes or variants that were previously unknown, and can provide large amounts of data in a short time. WGS can even predict the likelihood of genetic disorders, which can be mediated through lifestyle or environmental changes, to help in prevention of the occurrence of the disorders [37].
The second method is the targeted NGS. The targeted NGS is a type of NGS that focuses on specific genome areas, and scrutinize genome specific areas for deeper analysis. Therefore, it is faster and more cost-effective than WGS, but produce less data compared to WGS, as it analyse only selected specific areas [38].
The third method is the shotgun NGS, which is a technique to determine the sequence of all chromosomes and the whole genome by producing random DNA fragments with overlapping ends, followed by using a computer to assemble and order the fragments according to the overlapping ends into the whole genome. Shotgun NGS begins with the construction of a genome library. The genome library is produced by fragmenting the whole genome, the fragments are cloned into vectors, partially sorted and using a computer the data are collected to produce an entire genome sequences that consists of all fragments, which form known old sequences that are called contigs. “A contig is a set of overlapping DNA segments that together represent the consensus region of DNA” (cited from Yin and Zheng) [39]. Many contigs are often separated by unknown sequences. Unknown sequences can be determined either by hybridization protocols using known contig ends, or random PCR primers to
reveal the sequence of the gap region between known contigs. In hybridization protocol, the sequence of the gap between contigs is determined by filtering the hybridized clone library with a probe that is generated from the end of previously identified contigs. In random PCR primers’ method, the primers are annealed to the ends of two known contigs, and thus fill the gap and reveal the unknown sequence [39].
A special approach, which can be incorporated into the three methods are multiplexing, also called pooling, so that several samples can be processed simultaneously, to save costs and time. Multiplexing requires the addition of a barcode (index) to the sample so that they can be identified after being sorted. Samples used for hybridization catches can be duplicated after library preparation, but before target capture (enrichment). Samples used for sequential amplification must be transformed into libraries and enriched through individual PCR amplification before multiplexing to be sorted [39].
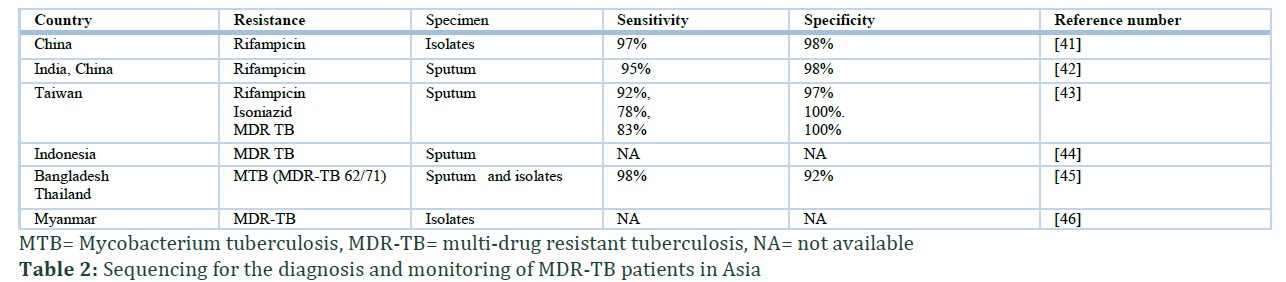
In a population-based multi-country study conducted by WHO using various NGS platforms (e.g. Illumina, Ion Torrent) and targeted methods, the results showed that NGS was very accurate in diagnosing MDR-TB from isolates that are cultured. 9 The study assessed various genes, including katG, promoter inhA and fabG to assess isoniazid, pncA to asses pyrazinamide, rpoB to asses rifampicin, gyrA and gyrB to asses ofloxacin and moxifloxacin, rrs to asses amikacin and capreomycin, and rrs and eis to asses kanamycin resistance. The study was conducted on 7.000 patients and found that there was good prediction accuracy of genetic sequences to show resistance to isoniazid, rifampicin, fluoroquinolones, and injections. This study strengths included the fact that the tested isolates came from a whole population of TB cases with a high TB or MDR-TB burden [10, 40]. Table 2 shows various studies that were conducted in Asian countries and used NGS and previous sequencing methods, which showed sensitivity and specificity of above 90% [41-46]. Most studies found that almost all specimens contained resistance to a minimum of two anti-TB drugs (isoniazid and rifampicin).
Many studies have been carried out using NGS method to test drug susceptibility of MTB, in which MTB susceptibility testing using NGS for clinical application gave faster results compared to the phenotypic testing.
Ko et al [47] studied eighteen susceptible strains with benign polymorphisms, to assess phenotypic and genotypic resistance patterns for isoniazid, rifampicin, ethambutol, and pyrazinamide. Thirty-nine variants were found in this study, and six mutations were regarded as causing resistance i.e.: katG c.C944G, rpoB c.C1349T, embB c.A916G and c.G918A, gyrA c.C269T and rpsL c.A128G. There were two main causes of isoniazid resistance, which were in katG and inha, but a most common variant, which was katG R463L, was regarded as a benign polymorphism. On the other hand, the most common resistant mutation, which was in S315T katG promoter area, was mostly found in Indonesia.The rifampicin rpoB mutation occurred in codon 407-533, and gave rise to S450L variant. For ethambutol resistance, mutation occurred in codon 306 of embB, while pyrazinamide resistance was due to two pncA mutations that might cause frame-shift mutations.
A recent study compared phenotypic testing with NGS Illumina platform to assess various TB drug resistance on overall isolates, and provided interpretations of mutations in katG, inha, fabG promoters, rpoB, pncA, gyrA, gyrB, rrs, and eis genes. The NGS was carried out by standard approaches to assess MTB mutations and provide drug resistance genetic sequencing data. The data was obtained by sorting all relevant genomes/genes to the target genome region of katG, inha, and fabG promoters for isoniazid, rpoB for rifampicin, pncA for pyrazinamide, and gyrA/gyrB for fluoroquinolones. The results on isolates from 7094 TB patients showed that overall sensitivity value of NGS to predict genetic resistance varies between various drugs. Good accuracy of sensitivity values was found for first-line drugs (katG promoter, inha, fabG, and rpoB) compared to second-line drug resistance, but lowest for detection of pyrazinamide resistance due to pncA mutation [10, 13].
Several studies showed that the main cause (96%) of rifampicin resistance was due to mutations, which occurred in the centre of a 81-base pair region in rpoB gene, and rpoB S531L was the most common mutation in clinical MTB isolates. In countries around the world, 40%-93% of all rifampicin resistant MTB isolates was due to rpoB S450L mutations. In addition, the most extensive rifampicin resistance was due to rpoB gene single amino acid mutation in codon 445 that changed His [CAC] to Tyr [TAC] [15-19].
Falzon et al [23] did a meta-analysis of individual MDR-TB patient data and concluded that the most common fluoroquinolone resistance was due to GCG90GTG and GAC94GGC mutations; where isolates with a GAC94GGC mutation usually showed a Minimum Concentration (MIC) that is higher compared to a GCG90GTG mutation.
A study concluded that NGS provided information that enable easier differentiation between various strains, and assessment of MTB genetic heterogeneity, by which MDR-MTB Beijing strain complex evolutionary pattern was revealed. Moreover, NGS showed more accuracy than other genotype methods, namely Mycobacterial interspersed repetitive unit- variable number tandem repeat (MIRU-VNTR) and Xpert MTB/RIF assay. So that the complex evolutionary patterns of Beijing MDR MTB strains in patients have been revealed using NGS [33].
Maningi et al study [21] using NGS showed that pncA gene mutation in Pyrazinamide resistance revealed additional diagnostic values that were accurate and might be considered in routine testing for MDR-TB management strategies. Eighty-eight isolates were sequenced using NGS Ion Torrent (PGM) platform, and 55 wild type and 33 pncA gene mutation containing isolates were found. PncA mutations in 33 of these isolates represent nucleotide substitution (missense mutations), deletions that cause amino acid substitution or frame shift mutation, and insertion. Using the NGS, additional mutations in 6 isolates were detected that were undetected in previous Sanger sequencing.
A study by Daum et al [17] in Ukraine used NGS to analyze MDR mutations in inhA, katG, and rpoB genomes and showed that all clinical isolates (100%) showed inhA wild type gene, among which 23% had mutation in inhA promoter region, while the most frequent mutation (89%) that caused isoniazid resistance occurred in katG S-315-T. For rifampicin resistance, 95% mutations occurred in rpoB ‘81-bp nucleus region’, which was rifampicin resistance determining region (RRDR), and most mutations occurred in positions 516 (D-516-V), 526 (H-526-Y or D) and 531 (S -531-L). Further, two rifampicin resistant isolates were isoniazid sensitive, and five rifampicin resistant strains showed heteroresistance, i.e.: two strains of S-531-S/F /L, which are rare mutations with one wild type and two mutant residues, two strains of S-531-S / L and one strain of H-526-H / D, where the three later strains were double mutants.
For amikacin and kanamycin resistance, rrs A1401G mutation, which caused high level resistance to both drugs, was the most common in Thailand (21 of 29 strains). This mutation showed cross-resistance with capreomycin. In addition, mutations in eis promoter caused low-level resistance to amikacin, which was found in five out of 29 strains that were kanamycin resistant [24]. Miotto et al [18] concluded that the mutations, which were associated with amikacin resistance, were only rrs A1401G and G1484T, and rrs mutations were associated with high MIC in a variety of different media.
Multi drug resistant is one of the global problems that occurs in almost all regions of the world, especially in Asian populations. In recent years, the NGS can be used as a rapid diagnostic tool in describing a patient's MDR-TB status. Almost all gene mutation, which causes resistance, can be found by this method. It is hoped that by using NGS, patient diagnosis and management will be more optimal, so that mortality due to MDR TB can be reduced.
ADVM conducted the literature search, chose the included studies, and drafted the tables and article. JAP added included studies, revised the tables and article, and added the discussion. The final version was approved by all authors.
The authors declare that there is no conflict of interest regarding the publication of this paper.
References ![]()
- Chakaya JM, Harries AD, Marks GB. Ending tuberculosis by 2030-Pipe dream or reality? International Journal of Infectious Diseases, (2020); 92S: S51-S54.
- WHO. Global tuberculosis report 2017. WHO, 2018. https://www.who.int/tb/publications/global_report/gtbr2017_main_text.pdf. Accessed on 20 May 2020.
- Walker T, Radcliffe J, Way H. Headington. DNA sequencing predicts 1st-line tuberculosis drug susceptibility. The New England Journal of Medicine, (2018); 379(15): 1403-1415.
- Zong Z, Huo F, Shi J, Jing W, Ma Y, Liang Q, et al. Relapse versus reinfection of recurrent tuberculosis patients in a national tuberculosis specialized hospital in Beijing, China. Frontiers in Microbiology, (2018); 9:1858.
- Gilpin C, Korobitsyn A, Weyer K. Current tools available for the diagnosis of drug-resistant tuberculosis. Therapeutic Advances in Infectious Diseases, (2016); 3(6):145-151.
- Nguyen TNA, Anton-Le Berre V, Bañuls AL, Nguyen TVA. Molecular diagnosis of drug-resistant tuberculosis; A literature review. Frontiers in Microbiology, (2019); 10: 794.
- Dlamini MT, Lessells R, Iketleng T, de Oliveira T. Whole genome sequencing for drug-resistant tuberculosis management in South Africa: What gaps would this address and what are the challenges to implementation?. Journal of Clinical Tuberculosis and Other Mycobacterial Diseases, (2019); 16: 100115.
- Hunter RL. The pathogenesis of tuberculosis: The early infiltrate of post-primary (adult pulmonary) tuberculosis: A distinct disease entity. Frontiers in Immunology, (2018); 9: 2108.
- McIvor A, Koornhof H, Kana BD. Relapse, re-infection and mixed infections in tuberculosis disease. Pathogens and Disease, (2017); 75: 3.
- WHO. The use of next-generation sequencing technologies for the detection of mutations associated with drug resistance in Mycobacterium tuberculosis complex: technical guide. WHO, 2018. https://apps.who.int/iris/handle/10665/274443. Accessed on 20 May 2020.
- WHO. Tuberculosis: Multidrug-resistant tuberculosis (MDR-TB). WHO, 2018. https://www.who.int/news-room/q-a-detail/what-is-multidrug-resistant-tuberculosis-(mdr-tb)-and-how-do-we-control-it. Accessed on 20 May 2020.
- CDC. Chapter 2 Transmission and Pathogenesis of Tuberculosis. https://www.cdc.gov/TB/education/corecurr/pdf/chapter2.pdf. Accessed on 20 May 2020.
- Nguyen L. Antibiotic resistance mechanisms in M. tuberculosis: An update. Archives of Toxicology, (2016); 90(7): 1585-1604.
- Song H, Sandie R, Wang Y, Andrade-Navarro MA, Niederweis M. Identification of outer membrane proteins of Mycobacterium tuberculosis. Tuberculosis (Edinb), (2008); 88(6): 526-544.
- Da Silva PEA, Palomin JC. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: classical and new drugs. Journal of Antimicrobial Chemotherapy, (2011); 66: 1417-1430.
- Zhang Y, Yew WW. Mechanisms of drug resistance in Mycobacterium tuberculosis: Update 2015. The International Journal of Tuberculosis and Lung Disease, (2015); 11: 1276-1289.
- Daum LT, Konstantynovska OS, Solodiankin OS, Liashenko OO, Poteiko PI, Bolotin VI, et al. Next-generation sequencing for characterizing drug resistance-conferring Mycobacterium tuberculosis genes from clinical isolates in the Ukraine. Journal of Clinical Microbiology, (2018); 56(6): e00009.
- Miotto P, Zhang Y, Cirillo DM, Yam WC. Drug resistance mechanisms and drug susceptibility testing for tuberculosis. Respirology, (2018); 23: 1098–1113.
- Palomino JC, Martin A. Drug resistance mechanisms in Mycobacterium tuberculosis. Antibiotics (Basel), (2014); 3(3): 317-340.
- Ando H, Akiyama TM, Watanabe S, Kirikae T. A silent mutation in mabA confers isoniazid resistant in Mycobacterium tuberculosis. Molecular Microbiology, (2014); 91 (3) : 538–547.
- Maningi NE, Daum L, Rodriguez J, Mphahlele M, Peters R, Fischer G, et al. Improved detection by next-generation sequencing of pyrazinamide resistance in Mycobacterium tuberculosis isolates. Journal of Clinical Microbiology (2015) ; 53(12): 3779-3783.
- Lin SYG, Desmond EP. Molecular diagnosis of tuberculosis and drug resistance. Clinics in Laboratory Medicine, (2014); 34: 297–314.
- Falzon D, Gandhi N, Migliori GB, Sotgiu G, Cox HS, Holtz TH, et al. Resistance to fluoroquinolones and second-line injectable drugs: Impact on multidrug-resistant TB outcomes. European Respiratory Journal, (2013); 42(1):156-168.
- Sowajassatakul A, Prammananan T, Chaiprasert A, Phunpruch S. Molecular characterization of amikacin, kanamycin and capreomycin resistance in M/XDR-TB strains isolated in Thailand. BMC Microbiology, (2014) ;14:165 .
- Jeanes C, O’Grady J. Diagnosing tuberculosis in the 21st century – Dawn of a genomics revolution? International Journal of Mycobacteriology, (2016); 5(4): 384-391.
- Dlamini MT, Lessells R, Iketleng T, de Oliveira T. Whole genome sequencing for drug-resistant tuberculosis management in South Africa: What gaps would this address and what are the challenges to implementation? Journal of Clinical Tuberculosis and Other Mycobacterial Diseases, (2019); 16: 100115.
- Cohen KA, Manson AL, Desjardins CA, Abeel T, Earl AE. Deciphering drug resistance in Mycobacterium tuberculosis using wholegenome sequencing: progress, promise, and challenges. Genome Medicine, (2019); 11: 45.
- McNerney R., Zignol M., Clark TG. Use of whole genome sequencing in surveillance of drug resistant tuberculosis. Expert Review of Anti-infective Therapy, (2018); 16(5):433-442.
- Dewey FE, Megan E, Grove MS, Pan C, Goldstein BA, Bernstein JA, et al. Clinical interpretation and implications of whole-genome sequencing. JAMA, (2014); 311(10): 1035-1045.
- Cirilo D, Blnaco M, Armaos A, Buness A, Avner P, Guttman M, et al. Quantitative predictions of protein interaction with long noncoding RNAs. Corespondence. Nature Methods, (2016); 14(1) : 5-6.
- Roetzer A, Roland Diel R, Kohl TA, Ru¨ckert C, Nu¨bel U, Blom J, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: A longitudinal molecular epidemiological study. PLOS Medicine, (2013); 10(2): e1001387 .
- Nikolayevskyy V, Trovato A, Broda A, Borroni E, Cirillo D, Drobniewski F. MIRU-VNTR genotyping of Mycobacterium tuberculosis strains using QIAxcel technology: A multicentre evaluation study. PLoS One, (2016);11(3): e0149435.
- Desikan S, Narayanan S. Genetic markers, genotyping methods & next generation sequencing in Mycobacterium tuberculosis. Indian Journal of Medical Research, (2015); 141(6): 761-774.
- Petersen LM, Martin IW, Moschetti WE, Kershaw CM, Tsongalis GJ. Third-generation sequencing in the clinical laboratory: Exploring the advantages and challenges of nanopore sequencing. Journal of Clinical Microbiology, (2019); 58(1): e01315-19.
- Mertes F, Elsharawy A, Sauer S, van Helvoort JMLM, van der Zaag PJ, Franke A, et al. Targeted enrichment of genomic DNA regions for next-generation sequencing. Briefings in Functional Genomics, (2011); 10(6): 374-386.
- Integrated DNA technologies. Targeted sequencing guide. https://go.idtdna.com/rs/400-UEU-432/images/IDT%20Targeted%20sequencing%20guide%20%281%29.pdf. Accessed on 20 May 2020.
- Clark DP, Pazdernik NJ. Genomic and system biology: Molecular biology. 2013; 2nd edition: e110-e117. Academic Cell.
- Yin R, Kwoh CK, Zheng J. Whole genome sequencing analysis: Encyclopedia of bioinformatics and computational biology. 2019; 3: 176-183. Elsevier.
- Zignol M, Cabibbe AM, Dean AS, Glaziou P, Alikhanova N, Ama C, et al. Genetic sequencing for surveillance of drug resistance in tuberculosis in highly endemic countries: A multi-country population-based surveillance study. The Lancet Infectious Diseases, (2018); 18(6): 675-683.
- Mbelele P, Mohamed S, Sauli E, Mpolya EA, Mfinanga SG, Addo K, et al. Meta‑narrative review of molecular methods for diagnosis and monitoring of multidrug‑resistant tuberculosis treatment in adults. International Journal of Mycobacteriology, (2018); 7(4): 299-309.
- Huang H, Zhang Y, Li S, Wang J, Chen J, Pan Z, et al. Rifampicin resistance and multidrug‑resistant tuberculosis detection using Xpert MTB/RIF in Wuhan, China: A retrospective study. Microb Microbial Drug Resistance, (2018); 24: 675‑679.
- Dorman SE, Schumacher SG, Alland D, Nabeta P, Armstrong DT, King B. Xpert MTB/RIF ultra for detection of Mycobacterium tuberculosis and rifampicin resistance: A prospective multicentre diagnostic accuracy study. The Lancet Infectious Diseases, (2018); 18: 76‑84.
- Lin HC, Perng CL, Lai YW, Lin FG, Chiang CJ, Lin HA, et al. Molecular screening of multidrug-resistance tuberculosis by a designated public health laboratory in Taiwan. European journal of clinical microbiology & infectious diseases, (2017) ; 36(12): 2431–2439.
- Chaidir L, Ruesen C, Dutilh BE, Ganiem AR, Andryani A, Apriani L, et al. Use of whole-genome sequencing to predict Mycobacterium tuberculosis drug resistance in Indonesia. Journal of Global Antimicrobial Resistance, (2019); 16: 170-177.
- Banu S, Pholwat S, Foongladda S, Chinli R, Boonlert D, Ferdous S, et al Performance of TaqMan array card to detect TB drug resistance on direct specimens. PloS one, (2017); 12(5), e0177167.
- San LL, Saw Aye K, Aye Thida N, Shwe MM, Fukushima Y, Gordom S, et al. Insight into multidrug Beijing Genotype Mycobacterium tuberculosis isolates in Myanmmar. International Journal Infection Diseases, (2018); 76: 109-119.
- Ko DH, Lee EJ, Lee EJ, Lee SK, Kim HS, Shin SY, et al. Application of next-generation sequencing to detect variants of drug-resistant Mycobacterium tuberculosis: Genotype-phenotype correlation. Annals of Clinical Microbiology and Antimicrobials, (2019); 18(1): 2.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0


