

Full Length Research Article
Protein Engineering of Endoglucanase CelR of Clostridium thermocellum for Enhanced Expression
Hafiz Muzzammel Rehman1, Hira Nasir2, Adnan Iqbal2, Syed Zawar Shah2*, Ammara Ahad2, Muhammad Umair Naseem2, Muhammad Sajjad3, Muhammad Waheed Akhtar4, Sajjad Ahmed4
Adv. life sci., vol. 6, no. 2, pp. 81-87, February 2019
*– Corresponding Author: Syed Zawar Shah (Email: syed.zawar01@gmail.com)
Authors' Affiliations
2- Centre of Excellence in Molecular Biology, University of the Punjab, Pakistan
3- University of Lahore, Pakistan
4. School of Biological Sciences, university of the Punjab, Lahore
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Enhanced production and improved properties of cellulases for a greater activity on plant biomass would rank amongst the top priorities for second-generation ethanol production. Based on the emergence of protein engineering as a cutting-edge technology for enhancing enzyme activity and expression level, the present study is aimed at the application of this technique to the major cellulosomal processing endoglucanase of C. thermocellum, CelR for refining enzyme characteristics.
Methods: The full-length native enzyme gene (CelR) and a truncated version without the docking domains at C-terminus (CelR-CB) were PCR amplified using gene specific primers. The amplified PCR products were T/A cloned in the vector pTZ57 R/T and transformed in E. coli DH5α. The cellulase genes from the confirmed transformed plasmids were sub-cloned in T7 promoter-based expression vector pET-28a and expression analysis was done in E. coli (DE3) BL21 codon Plus.
Results: An SDS PAGE analysis of both the CelR derivatives revealed that the truncated version i.e. CelR-CB showed a two-fold increase in expression level as compared to the full-length enzyme.
Conclusion: The increased expression level of CelR in E. coli coupled with its increased production therefore makes it a promising method for augmenting the recombinant enzyme production for potential applications.
Keywords: Cellulase, Clostridium thermocellum, Protein engineering, Biofuel production, Recombinant enzyme expression
Introduction![]()
Rising concerns about global climate change due to the combustion of fossil fuels has shifted scientific focus towards use of non-conventional (renewable) energy resources to sustain environment whilst fulfilling increased energy demands worldwide [1]. The structural polysaccharides comprising plant cell wall, mainly cellulose and hemicelluloses, represent the most abundant renewable, biological energy source in the world. These can be converted to industrially useful biofuels via the action of various enzymes. The use of plant biomass for second generation ethanol production would give several compensations such as minimizing the clash between land use for food and fuel production, providing inexpensive raw material and allowing less greenhouse gas emissions than those of other fuels [2].
Cellulose is a high molecular weight polymer consisting of several hundred to ten thousand beta-linked D-glucose units forming crystalline microfibrils, thus making it intrinsically resistant to microbial and enzymatic degradation known as “biomass recalcitrance” [3,4]. This inherent resistance of plants to hydrolysis can be overcome using consolidated bioprocessing- a promising strategy that combines the production of cellulases, substrate hydrolysis and fermentation in a single step, for sustainable energy production [2]. Cellulolytic microbes such as bacteria and fungi are exploited as potential producers of cellulases that are usually secreted as multiprotein complexes known as cellulosomes [5]. The three types of cellulases, classified on the basis of their mode of action, include cellobiohydrolases, endoglucanases and β-glucosidases, with the former two mainly involved in most of the cellulolytic activity. Cellobiohydrolases act by cleaving cellobiose residues successively from the ends of cellulose chains while endoglucanases cleave β-1,4-glycosidic bonds in the cellulose chains [6]. The two carboxylic groups present in the active site of the enzyme are involved in hydrolysis. The types of cellulases, with different specificities, work together in synergism. A large number of thermostable microbial cellulases find extensive applications in food, chemical, pharmaceutical, paper and waste treatment industries [7,8].
With attention drawn to cellulosic biofuels, scientists have laid much focus on the induction of more cellulase production by the native organism and development of an effective artificial cellulase system from cellulases of different sources, in non-natural hosts through recombinant DNA technology. Many cellulases of fungal and bacterial origin have been cloned into non-cellulase producing hosts, E. coli, S. cerevisiae and Schizosaccharomyces pombe with corresponding high-expression vectors [9-12]. The capacity to exploit cellulolytic enzymes through protein engineering, like those present inherently in the highly versatile cellulosome producing C. thermocellum, under optimal conditions holds immense prospects in the production of ethanol as an inexpensive renewable energy source. Four cellobiohydrolases CbhA, Ce1K, CelO, and CelS along with twelve endoglucanases CelA, CelB, CelD, CelE, CelF, CelG. CelH, CelJ, CelN, CelQ, CelR and CelT are produced by C. thermocellum that, unlike fungal celluloses, exhibit a significantly high activity on crystalline cellulose [12,13]. With recent advances in genetic engineering, Clostridium thermocellum has emerged as a potential candidate for consolidated bioprocessing due to its anaerobic, ethanologenic and thermophilic nature along with its amenability to co-culture; thereby offering a promising future in biofuels [14,15]. Cellulosic biofuels are not currently produced at a competitive level primarily due to high production costs of the cellulolytic enzymes.
The present study underscores the application of protein engineering technique to cellulase, CelR of Clostridium thermocellum for improving enzyme characteristics i.e. enhancement of expression level through truncation of native enzyme so as to reduce the cost of the enzyme. Endoglucanase R (CelR) is the major cellulosomal processing endoglucanase of C. thermocellum, having a catalytic domain of GH family 9 at N-terminus followed by a carbohydrate binding module of family 3 (CMB3) and dockering domains at the C-terminus. The full length native enzyme gene (CelR) and a truncated version without the dockering domains (CelR-CB) were successfully expressed in E.coli under pET expression system. The truncated version of CelR i.e. CelR-CB showed two fold increase in the expression level as compared to full length CelR; thereby making it a prospective approach to increase the recombinant enzyme production.
Methods
Reagents, Plasmids and Bacterial Strains
The plasmid pTZ57R/T was employed for gene cloning while pET28a was used to construct an expression vector. Gene manipulation was done using E. coli DH5α while E. coli BL21 (DE3) served as a host for pET expression system. The genomic DNA of C. thermocellum was purchased from ATCC (27405D). DNA purification was done using Fermentas DNA Extraction kit (K0513) while TA cloning was carried out using Fermentas InstaT/AcloneTM PCR Product Cloning kit (K1214) according to the manufacturers’ guidelines. Fermentas TransformAidTM Bacterial Transformation kit (K2711) was used for the preparation and transformation of competent cells of E. coli strains DH5α and BL21 (DE3). All the reagents used in experimental procedures were of analytical grade.
Primer Designing
According to the 2704 bp long nucleotide sequence of CelR of C. thermocellum (NCBI accession no. AJ585346) encoding a mature peptide, primer designing was done using software Primer 3.0 and FastPCR (Table 1) and their properties were checked by “Oligonucleotide Properties Calculator”. The gene’s restriction map was checked using NEBcutter and DNAmend. Restriction sites of NcoI and XhoI were added in the forward (CelR-F) and reverse (CelR-R) primers, respectively.
Amplification and TA cloning of native and truncated Cel-R gene
The DNA sequence of Cel-R and CelR-CB was PCR amplified using Taq DNA polymerase and the primers given in Table 1 as follows: denaturation at 95ºC, 40 s; annealing at 63.2ºC, 30 s; extension at 72ºC, 1 min 25 s for 30 cycles. Following amplification, the PCR products were analyzed on 1% agarose gel, the DNA was purified and TA cloned in pTZ57R/T. The recombinant plasmids (pTZ57R/T- cel-R; pTZ57R/T- celR-CB) were transformed into competent E. coli DH5α and cultured onto LB agar plates containing kanamycin (50μg/ml), isopropyl-β-D-thiogalactopyranoside (IPTG; 0.5mM) and 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal; 20μg/ml) at 37°C. Positive clones were confirmed by colony PCR and by blue-white screening. Ten well-formed white colonies were re-cultured on fresh LB agar media at 37°C (overnight). The cloned recombinant plasmids, confirmed by restriction digestion, were then purified from the culture medium by alkaline lysis with SDS and pellet dissolved in TE buffer (50μl). The isolated plasmids were then restriction digested with NcoI and XhoI for confirmation, checked on 1% agarose gel and stored at -20°C.
Construction of expression vectors
Plasmid pET-28a, that codes for a kanamycin resistance marker, was employed as a vector to direct the expression of cloned cel-R gene from its inducible T7 promoter as a His-tag fusion. The plasmid was linearized by restriction digestion with NcoI and XhoI that was checked by agarose gel electrophoresis. Purified restricted celR and celR-CB were then ligated into corresponding restriction sites of the linearized pET28a. The resulting ligation mixture was transformed into competent E. coli DH5α that were propagated at 37°C in LB agar medium having 50μg/ml kanamycin for the selection of transformants. Transformed E. coli DH5α colonies were subjected to plasmid isolation by alkaline lysis similar to the isolation of pTZ57R/celR (celR-CB) from E. coli DH5α. The isolated expression plasmids (pETCelR; pETcelR-CB) were confirmed by restriction digestion.
Expression of native and truncated Cel-R in shake flask
The confirmed expression constructs (pETCelR; pETcelR-CB) were finally transformed into competent E. coli BL21 (DE3) for cel-R expression. The bacterial host cells were cultured onto LB-agar plates containing kanamycin (50μg/ml) at 37°C. Positive clones were streaked onto fresh LB agar plates and allowed to incubate overnight at 37°C. Subsequently, a fresh clone of recombinant E. coli BL21 (DE3) containing pETCelR and pETcelR-CB respectively, was propagated in LB broth supplemented with 100μg/ml kanamycin at 37°C for 1-1.5 hours to an OD600nm of 0.5-0.8. Then 0.5mM of IPTG was added to induce the expression of cloned cellulase genes. The expression levels were systematically assessed by analyzing the samples at different induction times (0, 1, 2, 4, 6, 8 and 10 hours). pET28a vector transformed E. coli BL21 (DE3) cells were employed as a negative control.
Total cell protein analysis by SDS-PAGE
For evaluating protein expression in E. coli, 1 ml of the post-induction samples was individually centrifuged at 12,000 rpm for 1 minute. The respective cell pellets were resuspended in 75 μl of 1X phosphate buffered saline (PBS) followed by the addition of an equal volume of 2X SDS sample buffer to each sample. Further processing comprised of passing each sample through a 27-gauge needle 10-20 times and heating at 85°C for 5-10 minutes in a water bath. The samples were then subjected to 12% SDS-PAGE at 100V for 15 minutes. Detection of proteins was done by Coomassie brilliant blue R250 staining and protein fractions were assessed densitometrically.
Results![]()
Amplification and TA cloning of native and truncated Cel-R gene
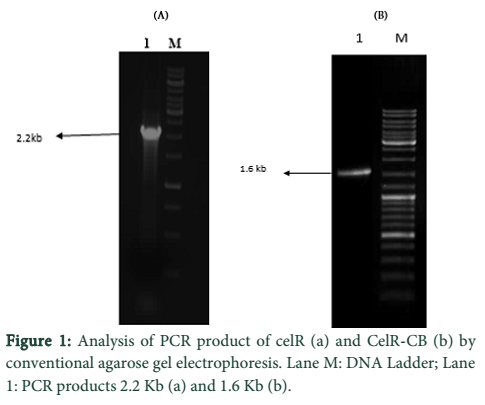
CelR was PCR amplified for 30 cycles from genomic DNA of C. thermocellum. Agarose gel analysis revealed successful amplification of both the native and truncated versions of CelR gene (Fig 1). PCR products were gel purified and cloned into pTZ57R/T. Transformation of E. coli DH5α cells with the recombinant plasmids (pTZ57R/T- cel-R; pTZ57R/T- celR-CB) was then carried out.
Transformation of E. coli DH5α with recombinant plasmids
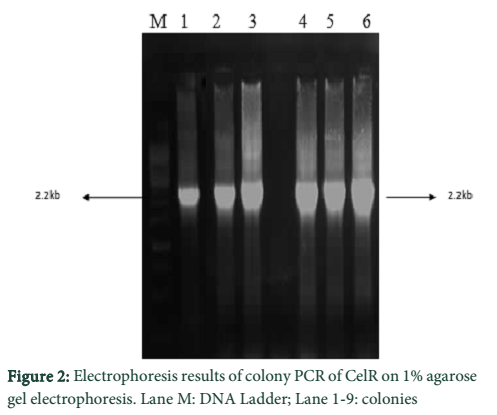
Following cloning of CelR into pTZ57R/T, transformation of competent E. coli DH5α cells were with the constructed recombinant plasmids (pTZ57R/T- cel-R; pTZ57R/T- celR-CB) was done. Positive clones were determined by blue-white colony screening and further confirmation was done by colony PCR.
Subcloning of CelR in expression vector pET-28a
The cloned recombinant plasmids were isolated from the culture medium and purified restricted celR and celR-CB were subcloned into linearized plasmid pET-28a at NcoI and XhoI sites. The expression plasmids (pETCelR; pETcelR-CB) transformed into E. coli DH5α were confirmed by restriction digestion.
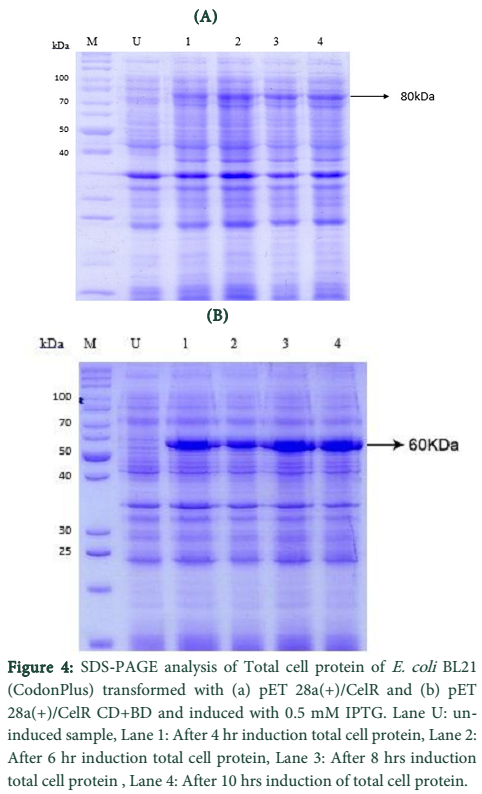
CelR expression in E. coli BL21 (DE3) and Total Cell Protein Analysis
The pET vector transformed E. coli BL21 (DE3) cells were induced with 0.5mM IPTG for directing cellulase gene expression. The expression levels were checked on 12% SDS-Polyacrylamide gel using SDS-PAGE.
Tables & Figures


Discussion![]()
In Clostridium thermocellum, a well-organized complex of cellulase and hemicellulase enzymes, referred to as cellulosome, is produced for hydrolyzing plant cell wall polysaccharides [16,17]. The use of plant biomass for second generation ethanol production endows several benefits including cutting-down dependence on fossil fuels and allowing less greenhouse gas emissions [18]. Production cost is one of the factor limiting cellulosic biofuel production which is mainly due to expense associated with the production of cellulolytic enzymes [19]. Protein engineering of cellulolytic enzymes under ideal conditions is regarded as an extremely promising approach in generating ethanol as a cost-effective renewable energy source [20]. Hence, this research was aimed at enhancing the expression of processive cellulase, CelR, from C. thermocellum through deletion of dockerin domain, located at the C-terminus of enzyme.
Endoglucanase R (CelR) is the chief cellulosomal processing endoglucanase of C. thermocellum, having an N-terminal catalytic domain, a carbohydrate binding module and a dockerin domain [21,22]. Both native (CelR) and truncated (CelR-CB) sequences were subjected to PCR amplification from the genomic DNA of C. thermocellum. PCR confirmation of successfully amplified DNA fragments was done by agarose gel analysis.
Optimization of MgCl2 concentration was done for the amplification of both enzyme sequences which indicated best amplification at 2.5 mM MgCl2. The DNA eluted from gel in each case was cloned into pTZ57R/T followed by transformation of E. coli DH5α cells with the constructs. The determination of positive clones was done through blue/white screening and colony PCR of white colonies. The PCR products of different sizes were obtained as expected; 2.2 kb for CelR and 1.6 kb for CelR-CB. The respective recombinant plasmids were isolated from a positive colony and further confirmed by restriction analysis, which indicated correctly restricted products.
The native type gene celR (encoding native enzyme) and its truncated derivative celR-CB (encoding the truncated enzyme), were subjected to sub-cloning from their respective pTZ-vectors into a T7-promoter based expression plasmid, pET28a(+) at NcoI and XhoI sites. Confirmation of the transformed E. coli DH5α colonies was done by colony PCR and restriction digestion of recombinant plasmids, pCelR and pCelR-CB with NcoI and XhoI, which revealed successful release of celR (2.2 Kb) and celR-CB (1.6 kb) from the respective recombinant plasmids. Transformation of E. coli BL21-CodonPlus (RIPL) cells with the recombinant expression plasmids was then carried out for expression analysis.
Expression of recombinant cellulase variants in E. coli BL21-CodonPlus (RIPL) was evaluated in LB medium using IPTG or lactose as an inducer [23]. Both CelR variants showed successful expression at their corresponding molecular weights in E. coli BL21-CodonPlus (RIPL) i.e. 80 kDa for native enzyme and 60 kDa for truncated version. Expression of CelR and CelR-CB expressed in E. coli BL21-CodonPlus (RIPL) was procured at levels of 12 and 125%, respectively of the total cell proteins as calculated densitometrically. Difference in heterologous expression of a recombinant protein in E. coli may be due to multiple factors; but most commonly it is regarded that larger the variant size, lower the expression level. Comparable recombinant expression patterns of derivatives of two main xylanases (XynC and XynZ) of C. thermocellum have been previously documented [24]. Expression of all the cellulase variants was obtained in the form of insoluble proteins in the cytoplasm as revealed by the SDS-PAGE analysis of sub-cellular fractions. No band of cellulase was observed in in soluble fraction in each case.
The study indicates that recombinant enzymes (i.e. CelR and CelR-CB) were successfully expressed in the media and, more importantly, CelR-CB construct exhibited over-expression due to deletion of dockerin domain at the C-terminal end. Further studies on soluble expression and refolding of enzyme are needed for functional characterization of engineered enzyme to divulge the effect of truncation of dockerin domain on enzyme function.
The authors declare that there is no conflict of interest regarding the publication of this paper.
References![]()
- Ben-Iwo J, Manovic V, Longhurst P. Biomass resources and biofuels potential for the production of transportation fuels in Nigeria. Renewable and Sustainable Energy Reviews, (2016); 63172-192.
- Lynd LR, Weimer PJ, Van Zyl WH, Pretorius IS. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev, (2002); 66(3): 506-577.
- Parsiegla G, Juy M, Reverbel‐Leroy C, Tardif C, Belaïch JP, et al. The crystal structure of the processive endocellulase CelF of Clostridium cellulolyticum in complex with a thiooligosaccharide inhibitor at 2.0 Å resolution. The EMBO Journal, (1998); 17(19): 5551-5562.
- Himmel ME, Ding S-Y, Johnson DK, Adney WS, Nimlos MR, et al. Biomass recalcitrance: engineering plants and enzymes for biofuels production. science, (2007); 315(5813): 804-807.
- Bayer EA, Chanzy H, Lamed R, Shoham Y. Cellulose, cellulases and cellulosomes. Current opinion in structural biology, (1998); 8(5): 548-557.
- Coutinho P. Carbohydrate-active enzymes: an integrated database approach. 1999. In Gilbert HJ, Davies G, Henrissat H, Svensson B (eds), Recent Advances in Carbohydrate Bioengineering. The Royal Society of Chemistry,Cambridge, pp. 3–12.
- Bhat M. Cellulases and related enzymes in biotechnology. Biotechnology advances, (2000); 18(5): 355-383.
- Haki G, Rakshit S. Developments in industrially important thermostable enzymes: a review. Bioresource technology, (2003); 89(1): 17-34.
- Okada H, Tada K, Sekiya T, Yokoyama K, Takahashi A, et al. Molecular characterization and heterologous expression of the gene encoding a low-molecular-mass endoglucanase from Trichoderma reesei QM9414. Appl Environ Microbiol, (1998); 64(2): 555-563.
- Nakatani F, Kawaguchi T, Takada G, Sumitani J-i, Moriyama Y, et al. Cloning and sequencing of an endoglucanase gene from Scopulariopsis brevicaulis TOF-1212, and its expression in Saccharomyces cerevisiae. Bioscience, biotechnology, and biochemistry, (2000); 64(6): 1238-1246.
- Voorhorst W, Eggen R, Luesink EJ, De Vos W. Characterization of the celB gene coding for beta-glucosidase from the hyperthermophilic archaeon Pyrococcus furiosus and its expression and site-directed mutation in Escherichia coli. Journal of Bacteriology, (1995); 177(24): 7105-7111.
- Zverlov V, Mahr S, Riedel K, Bronnenmeier K. Properties and gene structure of a bifunctional cellulolytic enzyme (CelA) from the extreme thermophile ‘Anaerocellum thermophilum’with separate glycosyl hydrolase family 9 and 48 catalytic domains. Microbiology, (1998); 144(2): 457-465.
- Guglielmi G, Béguin P. Cellulase and hemicellulase genes of Clostridium thermocellum from five independent collections contain few overlaps and are widely scattered across the chromosome. FEMS microbiology letters, (1998); 161(1): 209-215.
- Johnson EA, Madia A, Demain AL. Chemically defined minimal medium for growth of the anaerobic cellulolytic thermophile Clostridium thermocellum. Applied and Environmental Microbiology, (1981); 41(4): 1060.
- Mingardon F, Chanal A, López-Contreras AM, Dray C, Bayer EA, et al. Incorporation of fungal cellulases in bacterial minicellulosomes yields viable, synergistically acting cellulolytic complexes. Applied and Environmental Microbiology, (2007); 73(12): 3822-3832.
- Bayer EA, Belaich J-P, Shoham Y, Lamed R. The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annu Rev Microbiol, (2004); 58521-554.
- Najmudin S, Guerreiro CI, Ferreira LM, Romão MJ, Fontes CM, et al. Overexpression, purification and crystallization of the two C-terminal domains of the bifunctional cellulase ctCel9D-Cel44A from Clostridium thermocellum. Acta Crystallographica Section F: Structural Biology and Crystallization Communications, (2005); 61(12): 1043-1045.
- Jiang X, Wang Y, Xu L, Chen G, Wang L. Substrate binding interferes with active site conformational dynamics in endoglucanase Cel5A from Thermobifida fusca. Biochemical and biophysical research communications, (2017); 491(1): 236-240.
- Klein‐Marcuschamer D, Oleskowicz‐Popiel P, Simmons BA, Blanch HW. The challenge of enzyme cost in the production of lignocellulosic biofuels. Biotechnology and bioengineering, (2012); 109(4): 1083-1087.
- Wen F, Nair NU, Zhao H. Protein engineering in designing tailored enzymes and microorganisms for biofuels production. Current opinion in biotechnology, (2009); 20(4): 412-419.
- Gold ND, Martin VJ. Global view of the Clostridium thermocellum cellulosome revealed by quantitative proteomic analysis. Journal of bacteriology, (2007); 189(19): 6787-6795.
- Zverlov VV, Kellermann J, Schwarz WH. Functional subgenomics of Clostridium thermocellum cellulosomal genes: identification of the major catalytic components in the extracellular complex and detection of three new enzymes. Proteomics, (2005); 5(14): 3646-3653.
- Ramchuran S, Karlsson EN, Velut S, de Maré L, Hagander P, et al. Production of heterologous thermostable glycoside hydrolases and the presence of host-cell proteases in substrate limited fed-batch cultures of Escherichia coli BL21 (DE3). Applied microbiology and biotechnology, (2002); 60(4): 408-416.
- Sajjad M, Khan MIM, Akbar NS, Ahmad S, Ali I, et al. Enhanced expression and activity yields of Clostridium thermocellum xylanases without non-catalytic domains. Journal of biotechnology, (2010); 145(1): 38-42.
![]()
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0