

Full Length Research Article
Functional characterization of fifteen hundred transcripts from Ziarat juniper (Juniperus excelsa M.Bieb)
Humaira Abdul Wahid*, Muhammad Younas Khan Barozai, Muhammad Din
Adv. life sci., vol. 4, no. 1, pp. 20-26, November 2016
*– Corresponding Author: Humaira Abdul Wahid (Email: humairawahid@yahoo.com)
Authors' Affiliation
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Ziarat juniper (Juniperus excelsa M.Bieb) is an evergreen and dominant species of Balochistan juniper forests. This forest is providing many benefits to regional ecosystems and surrounding populations. No functional genomics study is reported for this important juniper plant. This research is aimed to characterize the Ziarat juniper functional genome based on the analyses of 1500 transcripts.
Methods: Total RNA from shoot of Juniperus excelsa was extracted and subjected for transcriptome sequencing using Illumina HiSeq 2000 with the service from Macrogen, Inc., South Korea. The Illumina sequenced data was subjected to bioinformatics analysis. Quality assessment and data filtration was performed for the removal of low-quality reads, ambiguous reads and adaptor sequences. The high-quality clean reads data was deposited in the Sequence Read Archive (SRA) at NCBI, and used for downstream processes. Fifteen hundred transcripts were randomly chosen and used for functional characterization.
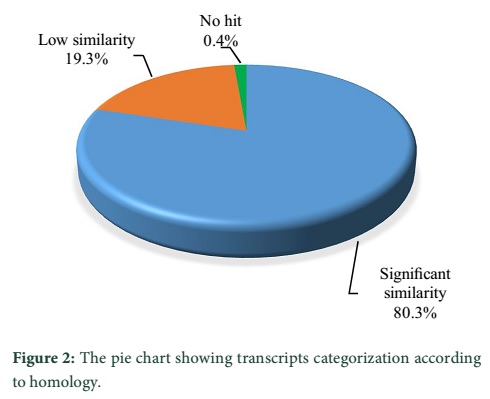
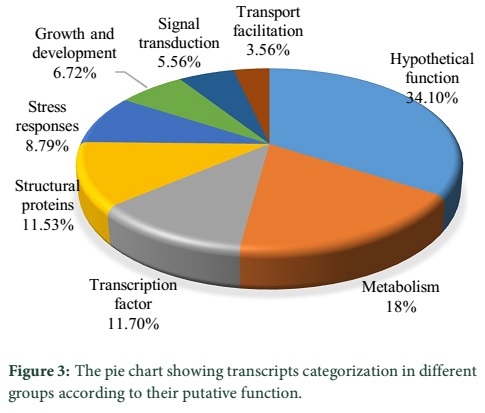
Results: As a result of homology search 80.3% transcripts showed significant similarities and were placed in significant similarities category, 19.3% transcripts showed low similarities and assigned to the ‘‘unclassified’’ category while 0.4% transcripts are defined as no hits. The functional characterization results showed that most (18%) of the transcripts are involved in metabolism, followed by 11.7% in transcription and 11.5% as structural protein. 8.8% transcripts are engaged in stress response, whereas the transcripts involved in growth and development constituted 6.7%. Transcripts involved in signal transduction represented 5.6%, while 3.5% facilitating transport and 34.1% are involved in hypothetical functions.
Conclusion: The functional annotation data produced in this study will be very useful for future functional genome analysis of Juniperus excelsa.
Keywords: Functional characterization, Illumina sequencing, Juniperus excels, transcriptome
Introduction
Juniper belongs to genus Juniperus, which consists of about 70 species occurring throughout the northern hemisphere of the world [1,2]. In Balochistan juniper forests spread between latitude 30°9´and 30°37´ N and longitude 67°11´ and 68°3´ E. in dry areas about 1200-3000m above sea level [3]. These juniper forests are pure tracks of Juniperus excelsa with one of the big block in Ziarat District. Juniperus excelsa is a large shrub or medium sized tree, spreads mainly in north-eastern Greece, southern Bulgaria, Turkey, Middle-East countries Syria and Lebanon, Saudi Arabia Iran, Pakistan, Oman and Caucasus Mountains at an altitude of 2000-4000m [4-6]. These plants are slow growing and can live up to 2000 years [7]. Juniperus excelsa forest of Ziarat have significance due to their old age, slow growth rate and provision of valuable services. In spite of a lot of significance presently no functional genomics study is reported for this plant.
In recent years, Next Generation Sequencing (NGS) has appeared as a high-throughput sequencing technology [8]. Various platforms use NGS, such as the 454 Roche Genome Sequencer, the SOLiDs ABI System, the Illumina Genome Analyzer, and showed to be efficient, powerful and cost-effective tools for advanced genomic research [9]. In the present study, Illumina sequencing HiSeq-2000 platform was used for transcriptome analysis of Juniperus excelsa. Fifteen hundred transcripts of Juniperus excelsa were randomly selected from Illumina sequenced data deposited in the Sequence Read Archive (SRA) at NCBI. The selected transcripts were used for functional characterization using BLASTn algorithm. To our knowledge, this is the first report on the characterization of the transcriptome of Juniperus excelsa and this new data-set will be a useful source for future genomic aspects such as to elucidate candidate genes for biotic and abiotic stress resistance, significant medicinal secondary metabolites and comparative genomics of Juniperus excelsa.
Methods
Plant Material
Samples (shoots) of Juniperus excelsa were collected from Sasnamana Valley, Ziarat, Balochistan, Pakistan. The collected samples were stored in air sealed polythene bags at -20°C for further analysis.
RNA Extraction
Total RNA extraction was performed by optimized RNA extraction method [10]. Briefly, 100 mg sample was ground in 1ml of preheated (65°C) total RNA extraction buffer 3% (w/v) CTAB, 3% (w/v) PVP, 100 mM Tris HCl (pH 8.0), 25 mM EDTA (pH 8.0), 2M NaCl, 2% SDS (w/v) and 2% 2-mercaptoethanol. The mixture was vigorously mixed and incubated at room temperature for 10 min with 2-3 times vortexing and centrifugation for 15 min at 15,500g at 4°C. Each sample was extracted twice by using an equal volume of chloroform:isoamyl alcohol (24:1), and phenol/chloroform/isoamyl alcohol (PCI) (25:24:1, v/v/v) at 15,500g for 20 min at 4°C. The resulting supernatant was carefully transferred into a new tube, mixed with 20ul of 5M sodium acetate and twice volume of 100% ethanol and was incubated overnight at -20°C for precipitation. After overnight precipitation RNA was collected by centrifugation at 15,500g at 4°C for 20 min. The pellet was washed with 70% ice cold ethanol. After air drying the RNA was dissolved in 80ul DEPC treated water and stored at -20°C until used. To check the integrity of total RNA samples were run on 1% agarose gel stained with ethidium bromide (EtBr), and visualized under UV light. Genova Nano spectrophotometer at an absorbance ratio of A260/230 and A260/280 nm was used to assess the quality and concentration of extracted total RNA. Quality analysis was performed through a Bioanalyzer (Agilent) at Macrogen, Inc., South Korea before deep sequencing.
Sequencing and Bioinformatics Analysis
Sequencing was performed through Illumina HiSeq-2000 platform with the service from Macrogen, Inc., South Korea according to the manufacturers’ protocols. The sequenced data was subjected to bioinformatics analysis, such as; data quality analysis by galaxy-FastQC read quality, removal of adaptor sequences by galaxy trimmomatic tool, homology search through BLAST algorithm and functional characterization by applying Gene-Ontology approaches. Sequenced reads were quality assessed with the quality assessment software FastQC (https://usegalaxy.org/). After quality assessment and data filtration reads with 94.67% at Quality score 20 were used for further analysis. The high quality reads produced have been deposited in the Sequence Read Archive (SRA) at NCBI (ncbi.nlm.nih.gov/sra) under accession number SRP082133.
Functional Annotation
Functional annotation was performed through BLASTn against the NCBI nonredundant (NR) DNA database (ftp://ftp.ncbi.nlm.nih.gov/ BLAST/db/). The transcripts with query coverage more than 50% were regarded as significant similarity. The transcripts with below 50% query coverage were deemed to have no significant similarity and were assigned to the ‘‘unclassified’’ category and transcripts with no similarity were defined as ‘‘no hits’’ [11-13]. The transcripts were further categorized on the basis of available literature into different functional categories that was metabolism, stress response, signal transduction, structural protein, transcription, transport facilitation, growth and development and hypothetical function [14,15].
Results
Total RNA is the starting material to perform all downstream analysis, so it should be in good quality and quantity. A modified method was applied for the extraction of high-quality total RNA from juniper shoots. Total RNA extraction showed 200.95µg/ml yield per sample, with absorbance ratio 2.13 at optical density 260/230 and 1.94 at optical density 260/280. The total RNA integrity was assessed by two distinct 28S and 18S rRNA bands (Figure 1).
The functional annotation showed that out of 1500 randomly selected transcripts 1205 transcripts (80.3%) were observed with significant similarities to the GenBank non-redundant database and placed in significant similarities category, 289 transcripts (19.3%) showed low similarities and assigned to the ‘‘unclassified’’ category. The remaining 6 (0.4%) transcripts with no similarity are defined as no hits (Figure 2).
Profiling of the putative functions presented that 18% (217) transcripts were engaged in metabolism, 11.7% (141) are playing role as transcription factors and 11.5% (139) were structural protein. 8.8% (106) transcripts were involved in stress responses, whereas 6.7% (81) were engaged in growth and development. Transcripts involved in signal transduction and transport represented 5.6% (67) and 3.6% (43) respectively. The 34.1% (411) transcripts were found with hypothetical protein nature (Figure 3).
Majority (18%) of the newly profiled transcripts were involved in the metabolism, such as ribulose bisphosphate carboxylase, cinnamoyl CoA reductase (CCR2. CCR7), starch synthase, hydroquinone glucosyltransferase, carbonic anhydrase, glycosyltransferase, zeta-carotene desaturase, fructose-bisphosphate aldolase. The 11.7% analyzed transcripts were engaged in transcription regulation including DEAD-box helicase 54, GTP-binding elongation factor, elongation factor 1-delta-like, MYB transcription factors (3R-MYB), zinc finger transcription factor and F-box/FBD/LRR-repeat protein. Transcripts engaged in transport activity were nitrate transporter, ATP-binding cassette (ABC) transporter, iron-inhibited ABC transporter, aromatic amino acid transporter and adenine nucleotide transporter.
The newly analyzed transcripts were also observed with roles in the process of cell signaling pathways like Rho small GTPase-activating proteins, receptor-like kinase, leucine-rich repeat receptor protein kinase (LRRK1) and cyclin-dependent kinase. Transcripts related with stress responses were heat shock proteins (HSP17, HSP70, HSP83, HSP88), galactinol synthase 1, endoplasmin homolog, Tobacco Mosaic Virus (TMV) resistance protein N-like.
Tables & Figures

Discussion
In the current research for functional characterization of Ziarat juniper transcripts, 80% transcripts were placed in significant similarities category. Similar homology categories were assigned to various plant by many researchers [16,17]. The rest 20% transcripts with no significant homologues in the NCBI-GenBank, representing the novel transcripts from Ziarat juniper that may have significant role in the maintenance of plant and human beneficial secondary metabolites.
Functional annotation of transcripts showed that maximum number of transcripts were involved in metabolism. Similar results were observed by Fei et al. [16] in leaves of mangrove plant. Among transcripts involved in metabolism, Fructose-bisphosphate aldolase (FBA) a key metabolic enzyme in glycolytic pathway of plants is also reported by Lu et al. [18] in Arabidopsis genome by using BLAST algorithms. Cinnamoyl CoA reductase (CCR) that catalyze the first specific step in lignin biosynthesis has been reported in the model plant Arabidopsis, Oryza and Populus [19-21]. CCR proteins have also been identified from and several economic plants such as Triticum aestivum and Zea mays [22,23]. Starch synthase an important enzymes for Starch biosynthesis in higher plants was also reported in the rice genome by Dian et al. [24] and Jiang et al. [25]. These identified transcripts with potential roles in metabolism would be helpful in understanding of metabolic pathways of juniper.
Numerous transcripts were found to be involved in transcription regulation like Zinc finger proteins and MYB transcription factors. The Zinc finger proteins (ZFPs) as significant transcription factors are widely reported in other plants [26,27]. Similarly, the MYB transcription factor is important for the regulation of several developmental and physiological processes in plants [28]. These functionally diverse transcription factors have been identified in Arabidopsis [29] and soybean [30]. Soler et al. [31] characterize the MYB family in Eucalyptus grandis genome, while Li et al. [32] identified and characterized MYB Transcription Factors in Pyrus bretschneideri. These transcripts represent a good information that could be utilized to improve the juniper forest based on transcription factors.
ATP-binding cassette (ABC) proteins that facilitate MgATP-energized transmembrane transport and regulate other transporters were reported by Rea [33] in Arabidopsis thaliana and Oryza sativa. Aromatic amino acids (AAAs) that function as precursors of several natural products and involved in synthesis of proteins, are also identified by Chen et al. [34] in Arabidopsis. For the growth and development of plant histone acetylation and deacetylation are very important [35]. Like our findings, involvement of transcripts with histone deacetylase activity is also reported in other plants by many researchers [36,37]. Findings regarding Glycine-rich cell wall structural proteins related transcripts were similar with the prior findings in bean and rice [38,39]. These findings would be of interest in future studies to determine the roles of juniper transcripts in growth and development, transport and as structural proteins.
Transcripts related to signal transduction such as Rho small GTPase-activating protein, have emerged as signal integrator and coordinators of a wide range of signaling pathways [40]. GTPases were discovered from a number of plant species such as barley and peach [41,42]. Genome sequencing studies showed that plant genomes encode a great number of receptor-like kinase (RLKs), For example, about 610 RLK coding sequences were reported in Arabidopsis genome [43]. Leucine-rich repeat receptor protein kinase (LRR-RKs) that controls a wide variety of developmental and defense-related processes was also reported in rice, poplar and soybean [44-46]. These findings may contribute in understanding of the mechanisms behind cell signaling pathways.
Heat shock proteins (HSPs) or chaperones play a crucial role in protecting plants against stress and assist in protein refolding. In our finding, many heat shock proteins are observed. According to Santhanagopalan et al. [47] HSPs function, not only during stress, but also during specific developmental stages in plants. In plants, many HSP proteins have been identified in different species [48,49]. Similarly, galactinol synthase catalyzes formation of galactinol associated with the responses to environmental stresses have been reported from Medicago falcata [50]. These findings regarding transcripts involved in cell signaling and stress responses would be significant candidates to understand the defense mechanism of Ziarat Juniper.
The profiling presented in this study provides the first overview of transcripts from the shoots of Ziarat juniper. This study analyzed 1500 transcripts with potential roles in metabolism, transcription, signal transduction, stress response, structural protein, growth and development and transport pathways of juniper plant. It is anticipated that the information obtained from this study such as; HSPs, ZFPs and LRR-RKs would be significant resources for future genomic studies and helpful to understand various mechanism under biotic and abiotic stresses in this plant.
Acknowledgment
This paper is a part of the research project (HEC-NRPU Project 20-1867/R&D/11) financed by the higher education commission (HEC) of Pakistan, Islamabad. The authors are highly thankful and acknowledge this financial support of the higher education commission (HEC) of Pakistan, Islamabad.
References
- Gulacti T, Ramazan E, Osman C, Candan J, Cennet C, Chai HB, Pezzuto JM. Diterpenes from the berries of Juniperus excels. Phytochemistry, (1999); 50(7): 1195-119.
- Farjon, A. World checklist and bibliography of conifers. Royal Botanic Gardens, Kew chemistry of natural compounds, (1998); 44(1): 2008.
- Rafi, M. (1965). Vegetation types of Balochistan Province. Pakistan Govt, Printing Press, Lahore, Pakistan.
- Weli AM, Jamila RK, AL-Hinai, Jawaher MA, Al-Mjrafi, Jawaher RS, Mohammad AH, Saeed S, Aktar MS. Effect of different polarities leaves crude extracts of Omani juniperus excels on antioxidant, antimicrobial and cytotoxic activities and their biochemical screening. Asian Pacific Journal of Reproduction, (2014); 3(3): 218-223.
- Khan M, Khan AU, Rehman NU, Zafar MA, Hazrat A, Gilani AH. Cardiovascular effects of Juniperus excelas, are mediated through multiple pathways. Clinical and Experimental Hypertension, (2012); 34: 209-216.
- Sela F, Karapandzova M, Stefkov G, Cvetkovikj I, Kulevanova S. Chemical composition and antimicrobial activity of essential oils of Juniperus excelsa Bieb. (Cupressaceae) grown in R. Macedonia. Pharmacognosy Research, (2015); 7(1): 74-80.
- Unlu M, Vardar-Unlu G, Vural N, Domnez E, Cakmak O. Composition and antimicrobial activity of Juniperus excelsa essential oil. Chemistry of Natural Compounds, (2008); 44: 129-31.
- Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One, (2012); 7 (2).
- Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics, (2008); 92(5): 255-64.
- Wahid HA, Barozai MYK, Din M. Optimization of total RNA extraction protocol from Ziarat Juniper (Juniperus excelsa M.Bieb). Pure and Applied Biology, (2015); 4(2):275-279.
- Bohnerta HJ, Ayoubid P, Borcherta C, Bressanc RA, Burnapd R, Cushmane JC, et al. A genomics approach towards salt stress tolerance. Plant Physiology and Biochemistry, (2001); 39: 295-311.
- Fu XH, Huang YL, Deng SL, Zhou RC, Yang GL, Ni XW, Li WJ, Shi SH. Construction of a SSH library of Aegiceras corniculatum under salt stress and expression analysis of four transcripts. Plant Science, (2005); 169: 147–154.
- Liu X, Huang J, Zhang Y, Lu Y. Identification of differentially expressed sequences in bud differentiation of oriental hybrid lily cultivar ‘Sorbonne’ via suppression subtractive hybridization. African Journal of Biotechnology, (2012); 11(66):12990-12997.
- Xu H, He X, Wang K, Chen L, Li K. Identification of early nitrate stress response genes in Spinach roots by suppression subtractive hybridization. Plant Molecular Biology Reporter, (2012); 30: 633-642.
- Roohie RK, Umesha S. Identification of genes associated with black rot resistance in cabbage through suppression subtractive hybridization. Springer 3 Biotech, (2015); 5:1089-1100.
- Fei J, Wang Y, Jiang Z, Cheng H, Zhang J. Identification of cold tolerance genes from leaves of mangrove plant Kandelia obovata by suppression subtractive hybridization. Ecotoxicology, (2015); 24:1686-1696.
- Tian j, Belanger F, Huang B. Identification of heat stress-responsive genes in heat-adapted thermal Agrostis scabra by suppression subtractive hybridization. Journal of Plant Physiology, (2009); 166(6): 588-601.
- Lu W, Tang X, Huo Y, Xu R, Qi S, Huang J, Zheng C, Wu CA. Identification and characterization of fructose 1,6-bisphosphate aldolase genes in Arabidopsis reveal a gene family with diverse responses to abiotic stresses. Gene, (2012)15; 503(1): 65-74.
- Costa MA, Collins RE, Anterola AM, Cochrane FC, Davin LB, Lewis NG. An in silico assessment of gene function and organization of the phenylpropanoid pathway metabolic networks in Arabidopsis thaliana and limitations thereof, Phytochemistry, (2003); 64:1097-1112.
- Kawasaki T, Koita H, Nakatsubo T, Hasegawa K, Wakabayashi K, Takahashi H, Umemura K, Umezawa T, Shimamoto K. Cinnamoyl-CoA reductase, a key enzyme in lignin biosynthesis, is an effector of small GTPase Rac in defense signaling in rice. Proceeding of National Academy of Science, (2006); 103: 230-235.
- Li L, Cheng X, Lu S, Nakatsubo T, Umezawa T, Chiang VL. Clarification of cinnamoyl co-enzyme A reductase catalysis in monolignol biosynthesis of Aspen. Plant Cell Physiology, (2005); 46:1073-1082.
- Ma QH. Characterization of a cinnamoyl-CoA reductase that is associated with stem development in wheat. Journal of Experimental Botany, (2007); 58: 2011-2021.
- Andersen JR, Zein I, Wenzel G, Darnhofer B, Eder J, Ouzunova M, Lubberstedt T. Characterization of phenylpropanoid pathway genes within European maize (Zea mays L.) inbreds. BMC Plant Biology, (2008); 8: 2.
- Dian WM, Jiang HW, Chen QS, Liu FY, Wu P. Cloning and characterization of the granule-bound starch synthase II gene in rice: gene expression is regulated by the nitrogen level, sugar and circadian rhythm. Planta, (2003); 218: 261-268.
- Jiang HW, Dian WM, Liu FY, Wu P. Molecular cloning and expression analysis of three genes encoding starch synthase II in rice. Planta, (2004); 218: 1062-1070.
- Barozai MYK, Wahid HA. In-silico identification and characterization of cumulative abiotic stress responding genes in potato (Solanum tuberosum L.). Pakistan Journal of Botany, (2012) 44: 57-69.\
- Cheuk A, Houde M. Genome wide identification of C1‑2i zinc finger proteins and their response to abiotic stress in hexaploid wheat. Molecular Genet Genomics, (2016); 291:873-890.\
- Hichri I, Barrieu F, Bogs J, Kappel C, Delrot S, Lauvergeat V.Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. Journal of Experimental Botany, (2011); 62(8): 2465-2483.
- Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in Arabidopsis. Trends in Plant Sciences, (2010); 15(10): 573-581.
- Du H, Yang SS, Feng BR, Liu L, Tang YX, et al. Genome-wide analysis of the MYB transcription factor superfamily in soybean. BMC Plant Biology, (2012); 12: 106.
- Soler M, Leal E, Camargo O, Carocha V, Cassan-Wang H, Clemente HS, Savelli B, Charles A,Jorge A, Paiva P, Myburg AA, Grima-Pettenati J. The Eucalyptus grandis R2R3-MYB transcription factor family: evidence for woody growth-related evolution and function. New Phytologist, (2015); 206(4): 1364-1377.
- Li X, Xue C, Li J, Qiao X, Li L, Yu L, Huang Y. Genome-Wide Identification, Evolution and Functional Divergence of MYB Transcription Factors in Chinese White Pear (Pyrus bretschneideri) Plant and Cell Physiology, (2016); 57(4): 824-847.
- Rea PA. Plant ATP-Binding Cassette Transporters. Plant Biology, (2007); 58: 347-375.
- Chen L, Ortiz-Lopez A, Jung A, Bush DR. ANT1, an Aromatic and Neutral Amino Acid Transporter in Arabidopsis. American Society of Plant Physiologists, Plant Physiology, (2001); 125 (4): 1813-1820.
- Hollender C, Liu Z. Histone Deacetylase Genes in Arabidopsis Development. Journal of Integrative Plant Biology, (2008); 50 (7): 875-885.
- Wu K, Tian L, Malik K, Brown D, Miki B. Functional analysis of HD2 histone deacetylase homologues in Arabidopsis thaliana. Plant Journal, (2000); 22: 19-27.
- Dangl M, Brosch G, Haas H, Loidl P, Lusser A. Comparative analysis of HD2 type histone deacetylases in higher plants. Planta, (2001); 213: 280-285.
- Keller B, Sauer N, Lamb CJ. Glycine-rich cell wall proteins in bean: gene structure and association of the protein with the vascular system. The EMBO Journal, (1988); 7(12): 3625-3633.
- Lei M, Wu R. A novel glycine-rich cell wall protein gene in rice. Plant Molecular Biology, (1991); 16 (2): 187-198.
- Fu Y, Yang Z. Rop GTPase: a master switch of cell polarity development in plants. Trends Plant Science, (2001); 6: 545-547.
- Schultheiss H, Dechert C, Kogel K, Huckelhoven R. Functional analysis of barley RAC/ROP G-protein family members in susceptibility to the powdery mildew fungus. The Plant Journal, (2003); 36(5): 589-601.
- Falchi R, Cipriani G, Marrazzo T, Nonis A, Vizzotto G, Ruperti B. Identification and differential expression dynamics of peach small GTPases encoding genes during fruit development and ripening. Journal of Experimental Botany, (2010); 61(10): 2829-2842.
- Shiu SH, Bleecker AB. Receptor-like kinase from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proceeding of National Academy of Science, (2001); 98:10763-10768.
- Sun XL, Wang GL. Genome-wide identification, characterization and phylogenetic analysis of the rice LRR-kinases. PLoS ONE, (2011); 6(3):160-179.
- Zan YJ, Ji Y, Zhang Y, Yang SH, Song YJ, Wang JH. Genome-wide identification, characterization and expression analysis of populus leucine-rich repeat receptor-like protein kinase genes. BMC Genomics, (2013); 14:318.
- Zhou F, Guo Y, Qiu L. Genome-wide identification and evolutionary analysis of leucine-rich repeat receptor-like protein kinase genes in soybean. BMC Plant Biology, (2016) 16:58.
- Santhanagopalan I, Basha E, Ballard KN , Bopp NE, Vierling E. Model Chaperones: Small Heat Shock Proteins from Plants. The series Heat Shock Proteins, (2015); 8: 119-153.
- Barozai MYK, Husnain T. Identification of biotic and abiotic stress up-regulated ESTs in Gossypium arboreum. Molecular Biology Reports, (2012) 39 (2): 1011-1018.
- Wang R, Zhang Y, Kieffer M, Yu H, Kepinski S, Estelle M. HSP90 regulates temperature-dependent seedling growth in Arabidopsis by stabilizing the auxin co-receptor F-box protein TIR1. Natural communication, (2016); DOI: 10.1038/ncomms10269.
- Zhuo C, Wang T, Lu S, Zhao Y, Li X, Guo Z. A cold responsive galactinol synthase gene from Medicago falcata is induced by myo-inositol and confers multiple tolerances to abiotic stresses. Physiologia Plantarum, (2013); 149(1):67-78.