

Short Communication:
Genetic diversity and evolutionary analysis of Citrus Tristeza Virus p20 gene in Pakistan: insights into the spread and epidemiology
Noor-Ul-Huda Ghori*1, Muhammad Qasim Hayat1, Salvatore Davino2,3
Adv. life sci., vol. 3, no. 3, pp. 75-82, May 2016
*- Corresponding Author: Noor-Ul-Huda Ghori, (Email: hudaghori@gmail.com)
Authors' Affiliation
2- Department of Agricultural and Forest Science, University of Palermo, Viale delle Scienze Ed. 5, 90123 Palermo – Italy
3- Euro-Mediterranean Institute of Science and Technology (IEMEST), Via E. Amari 123, 90139 Palermo – Italy
Abstract![]()
Introduction
Methods
Results
Discussion
Supplementary Data
References
Abstract
Background: Citrus tristeza virus (CTV) is a widespread disease and the most destruction causing agent of citrus. Pakistan is ranked amongst the top ten citrus producing countries around the globe and it contributes about 2% to its foreign exchange earnings. Based on this assumption it is very important to monitor and determine the evolutionary forces and the phylogeography of Pakistani CTV population.
Methods: A total of 49 sequences of p20 gene from Pakistan were phylogenetically compared with CTV sequences worldwide. These sequences were analyzed for their genetic diversity and evolution using a Bayesian Probability approach and predicted secondary structure.
Results: Phylogenetic analysis using Bayesian probability inference and predicted secondary structures diversity of CTV indicated that Pakistani isolates were not diverse from global isolates. Lineage analysis showed that CTV was introduced in Pakistan in three individual events from various parts of the world. After that CTV dispersed in Pakistan via vector transmission or by use of infected propagating material by local farmers.
Conclusions: Our study confirmed multiple introductions of CTV in Pakistan and also confirmed the dissemination of CTV within Pakistan. This study also shows that the mutations are present in the predicted secondary structure of the p20 protein, however, it is not known if it affects the pathogenicity of the virus.
Keywords: CTV, Bayesian probability, phylogenetic analysis, p20 gene
Introduction
Citrus is rated among the most widely grown fruit crops in the tropical and subtropical regions of the world. Brazil, China, USA, Mexico, Argentina, Pakistan, India, Spain, Italy, Egypt, Turkey and Japan are the major citrus producing areas [1]. Pakistan being the major citrus producer contributes about 2% to the world’s citrus market of which 96% comes from Punjab [2]. Citrus is widely distributed in Pakistan consuming about 194.5 thousand hectares of land providing annual citrus yield of about 1982.2 thousand tons during 2010–11 [3]. One of the most destructive diseases that affect citrus worldwide is caused by Citrus tristeza virus (CTV).
CTV belongs to family Closteroviridae; genus Closterovirus. The virion is flexuous, filamentous having about ~20 kb ssRNA genome (Fig. 1). It consists of two coat proteins namely major coat protein (CP, coding for p25 gene) accounting for 95% of the virus and minor coat protein (CPm, coding for p27 gene) contributing 5% of the virus length [4]. The genome has 12 open reading frames (ORFs) encoding about 19 proteins and is considered as one of the largest RNA viruses known [5]. The 5′ terminal consists of two ORFs covering approximately half of the genome length, namely ORF1a and ORF1b that encodes for helicase, methyltransferase and RNA polymerase proteins. The other ten ORFs are present at 3′ end and are primarily involved in encoding movement proteins [6]. They are expressed through sub-genomic RNA (sgRNA) and include p33, p6, p65, p61, p27, p25, p18, p13, p20 and p23, p25, p27, p65 (homolog of Heat Shock Protein) and p61 are involved in assembly of virions [7]; p6 is a hydrophobic protein and has a hypothetic role in virus movement [6, 8]. p25, p20 and p23 are related with mechanism of suppressors of RNA silencing [9]; and finally p33, p13 and p18 play role in extending the virus host range. RNA silencing is a natural defence system stimulated in the plants upon viral insemination and is known as a conserved endogenous antiviral mechanism in plants [10]. During viral replication, viral double stranded RNAs (dSRNA) are produced which are ultimately recognized and degraded by RNA silencing proteins produced by the plants. There have been many studies conducted to construct artificial microRNA and complementary hairpin RNAs to generate resistant plants against viral diseases. The p20 gene (549nt) is the major component of CTV that accumulates in the amorphous inclusion bodies in addition to obstructing intra and intercellular RNA silencing by host plants.
CTV infects all the citrus species, cultivars and hybrids inducing mild to severe infections [11]. The virus is transmitted in a semi persistent manner by aphids, including Aphis gossypii, A. spiraecola, A. citricola, Dactynotus jacae, Myzus persicae, Toxoptera aurantii and the most threatening T. citricida [12]. Grafting using infected root stock also causes the spread of this virus. The virus is limited to phloem and its strains vary in symptom severity ranging from none to devastating effects [13]. The appearance of symptoms depend on various factors such as the vector specie, citrus cultivar, the time of infection or the rootstock used and the virus isolate that cause the disease. CTV causes three distinct syndromes: tristeza, stem pitting and seedling yellows. Tristeza causes decline of varieties grafted on sour orange; stem pitting is the most destructive syndrome related to CTV disease and brings tree to death in a short time; seedling yellows is usually observed by biological indexing, and in the nursery of sour orange but rarely in the field [14]. In Pakistan it was believed that only mild strains of CTV are present however, the virus has been detected in citrus species found in Punjab and KPK where it is a serious threat especially for the sour orange [11, 15].
CTV is the only Closterovirus specie that exists as multiple, phylogenetically similar strains [14]. Although it belongs to RNA viruses having a slow evolutionary rate [13], significant amount of variation has been observed in the genome of its variants classifying them into six bio-groups. These bio-groups includes VT, T36, T30, HA16-5, B165 and resistance breaking group [16, 17, 5]. It has been reported that CTV genomes of the variants are highly conserved at their 3′ end having nucleotide identity ≥ 90% but have great diversity generally due to recombination seen at the 5′ end and in ORF 1a and 1b genes [18-21]. This diversity is responsible for variability in disease symptoms worldwide [22].
Phylogenies are now widely used to infer the origin and spread of viral infections and their migration patterns [23, 24]. The study of evolutionary relationship of CTV is an important zone that needs to be explored and there is insufficient evolutionary data on CTV variants in Pakistan. In the current work, 49 sequences of p20 gene representatives from Pakistan and major citrus producers around the world were analysed using maximum likelihood and Bayesian evolutionary methods to construct evolutionary relationship between different variants and their distribution across the world and to determine the origin of Pakistani strains. Moreover, diversity of predicted secondary structure of p20 gene was analysed to see if the protein structure is as diverse as the nucleotide sequences of the viruses.
Methods
Taxon sampling
A total of three hundred twenty-five sequences of p20 gene of CTV were retrieved from Genbank (http://www.ncbi.nlm.nih.gov/) including sequences from Pakistan, Argentina, China, Egypt, Greece, India, Israel, Italy, Japan, Mexico, South Africa, Taiwan, Spain, Thailand and USA. All sequences were renamed as follows: Accession number: Country: Date. The criteria for selection of the sequences was (a) All sequences should be non- recombinants (b) The country of origin should be clearly established (c) isolation or publication date should be clearly established in the literature. All sequences with incomplete information were not selected for analysis.
Alignment and sequence processing
Due to large number of data a sequence selection was carried out. Sequences retrieved were grouped in sets representing their countries and aligned by multiple sequence alignment using Geneious version 6.1.8 (http://www.geneious.com). The aligned sequences were manually inspected and cropped. Neighbor- joining (NJ) method was used to perform phylogenetic analysis for each group using Geneious version 6.1.8 HKY was devised as best fit model by using J model test version 2.1.3 [25] according to Akaike information criterion for the data. The data was filtered for all countries except Pakistan by taking a representative isolate from each clade in the phylogenetic tree respective of each country (Fig. 2). All sequences of CTV p20 gene from Pakistan were included in the study for in depth analysis. The final number of sequences in the filtered set was 49 sequences (Table 1 in supplementary data) which were realigned and a data matrix was created by manual inspection and missing bases were filled with consensus sequence. The final alignment was used for subsequent phylogenetic and protein analysis.
Phylogenetic analysis
To construct the phylogenetic tree, 50 sequences (49 representative sequences, 1 out-group of Barley yellow stunt virus) were used to conduct Bayesian analysis Markov chain Monte Carlo (MCMC) approach using Mr. Bayes version 2.0.9 [26] plugin in Geneious version 6.1.8. The parameters of analysis were set at default (Rate variation = gamma, gamma categories = 4, Chain length = 1,100,000, heated chains = 4, heated chain temp = 0.2, subsampling frequency = 200, burn in length = 100,000, random seed = 29,600, unconstrained branch length: exponential = (10), shape parameter exponential = (10). The HKY + I + G substitution model was found to be best fit for the data according to Akaike information criterion in Jmodel test version 2.1.3 [25].
Construction of secondary structures
Secondary structures of p20 gene from all isolates were predicted using Geneious version 6.1.8. The first six conserved codons were deleted and sixth codon was considered the first codon for translation.
Results
Phylogenetic analysis
The phylogenetic investigation and analysis using Bayesian approach of 14 CTV p20 Pakistani isolates (from GenBank; Table 1 provided in supplementary data) and selected 35 CTV p20 worldwide isolates (from GenBank; Table 1 in supplementary data) gave six main clades with a high Bayesian probability support (Fig. 3). It can be inferred from the established clades that Pakistani isolates are polyphyletic rather than being monophyletic as expected from a single introduction event. Pakistani isolates were distributed in three individual clades (Clade IV, V and VI). Interestingly, they appeared together in a clade frequently; and seldom appeared with different countries in sub clades accounting for the local spread of the genotype likely via infected root stock by local farmers. The recurrence of Pakistani sequences together also shows a shared transmission route of the virus that is responsible for the ongoing and sustained transmission of it.
Clade IV composed of two isolates from Pakistan clustered with South Africa, Greece, India, Argentina and Israel. Whereas, Clade V showed 11 Pakistani sequences emerging with Italy, India, Argentina and Egypt. In clade VI a unique sequence from Pakistan appeared close to China amongst Argentina, USA, Italy, Spain, Mexico, Greece, Thailand and Taiwan. Each clade represents a separate introduction of CTV virus in Pakistan however, a close relationship has been observed between p20 genes among isolates within each clade as indicated by short branch lengths. The origin of CTV virus is difficult to track from the tree due to slow evolutionary rate of some genotypes [27, 13, 28].
Diversity in predicted secondary structure of CTV p20 protein
Secondary structure was predicted after translation of aligned CTV p20 representative sequences (Fig. 4) to check diversity of Pakistani CTV isolates p20 protein. The secondary structure of p20 protein was found to be quit conserved among Pakistani isolates with few differences. The residues marked in Figure 4 indicates that α helix was replaced by β strand and α helix was shortened with insertion of a β strand. Insertions of coil and turns were also noticed. Mutations in all proteins were detected at the start and end codons of the protein with almost conserved middle region. Surprisingly, the unique sequence clustered in clade VI was also found to be conserved among the Pakistani sequences and no significant changes were observed in secondary structure of p20 protein as compared to other Pakistani isolates. It is concluded that although changes at nucleotide level are found however, these mutations do not have significant effect on the secondary structure of the protein which largely remains unchanged among various viral strains. Moreover, conserved and variable regions of the p20 gene provide its ancestral relationships and divergence respectively.
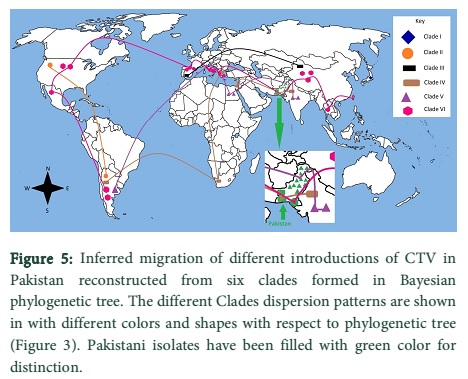
Migration patterns of CTV p20 gene worldwide
The migration patterns of CTV p20 gene were determined from Bayesian inference phylogeny analysis and represented through map (Fig. 5). Each introduction of CTV isolate determined from the phylogenetic tree was considered separately. Isolates of Clade VI were found to be dispersed in many diverse regions of the world including a unique sequence in Pakistan. Clade V was concentrated mostly in Pakistan. Interestingly, the other sequences observed in this clade were not from the close neighbouring countries of Pakistan with exception of India; instead they were from distant countries such as Argentina. Clade II was centred in South and North America whereas, Clade I isolate were only found in a single location indicating absence of dispersal of this lineage worldwide. The dispersal of Clade III lineage was observed to be restricted to two distant sites and was not dispersed rapidly.
Figures
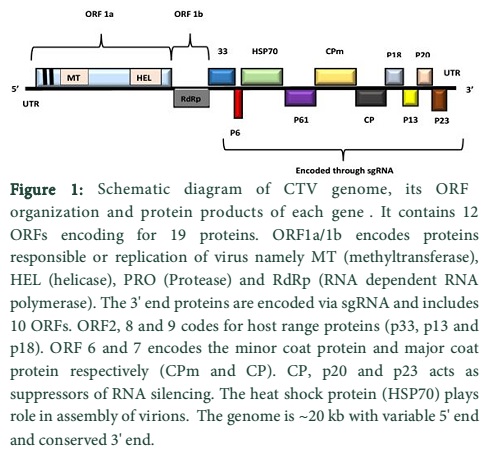
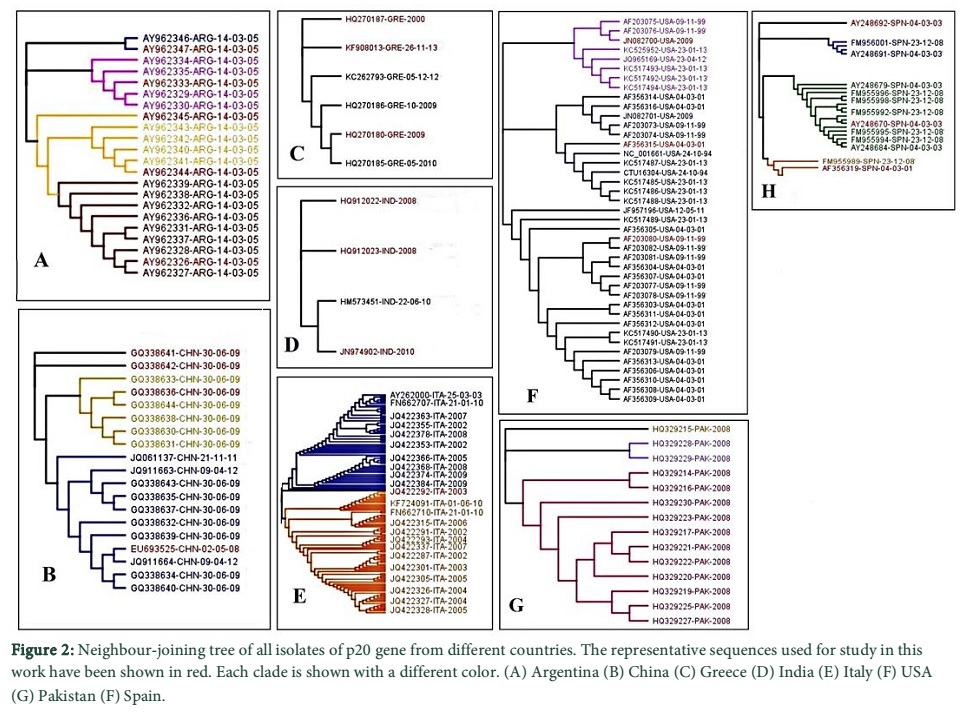
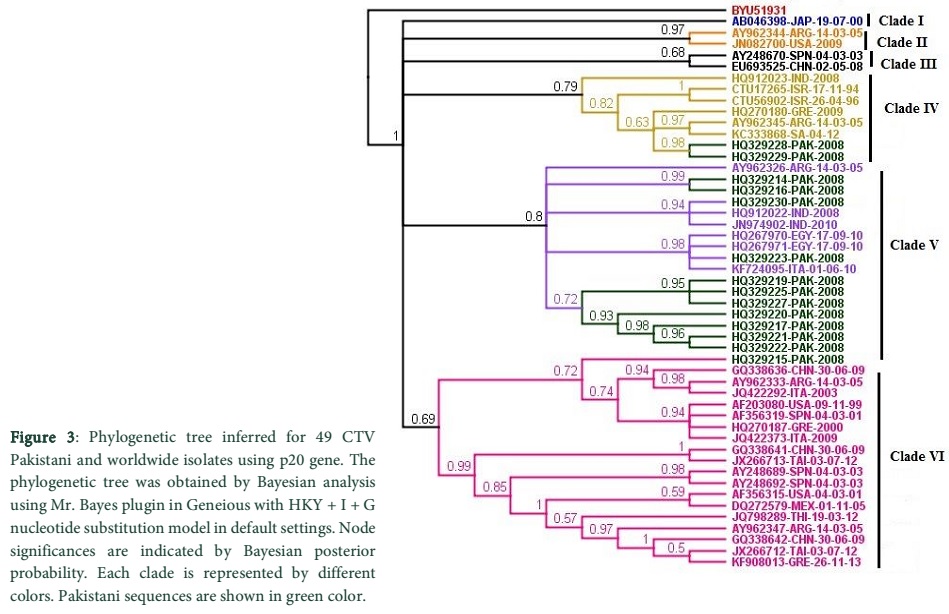
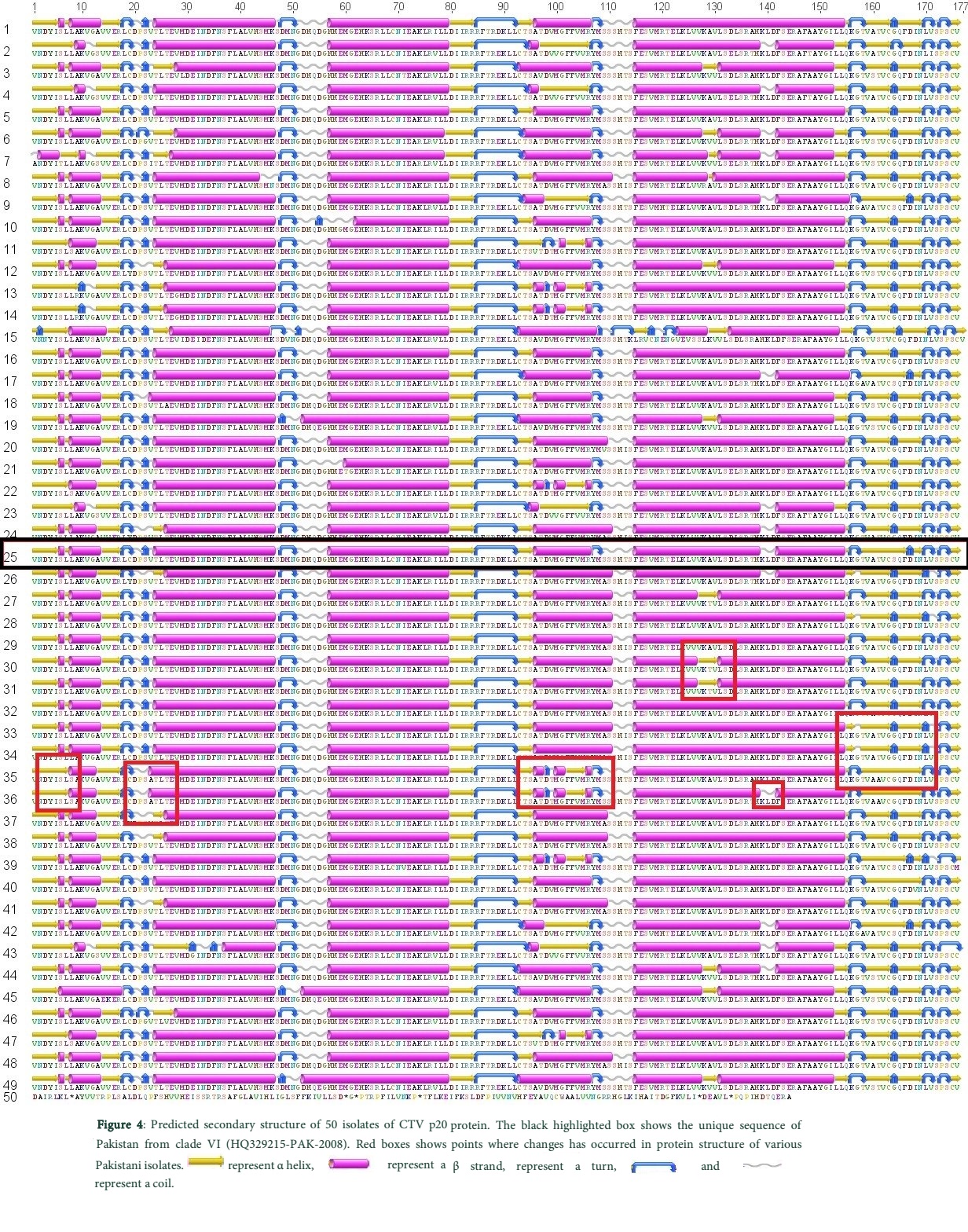
Discussion
In the current study, the origin and evolution of CTV p20 isolates from Pakistan in relevance to isolates worldwide with potential CTV threat has been examined by a phylogenetic approach. Nucleotide sequence analysis is a precise and accurate method for differentiation and estimation of genetic variation of CTV isolates [28]. The diversity of secondary structure and the study of changes brought about in the predicted secondary structure of p20 protein of Pakistani isolates have been investigated. The Bayesian probability phylogenetic analysis showed six clades consisting of isolates from Pakistan and other geographical regions. Pakistani isolates were however, found to be clustered in only three clades which confirmed that CTV has been introduced in Pakistani citrus variety through three independent events. This demonstrated that, CTV infections are polyphyletic in nature, as reported by [29] in phylogenetic analysis in Sicily, Italy. Based on phylogenetic analysis it has been observed that CTV isolates are subjected to divergence and are distributed in various clades (bio-groups) (Fig. 3) and few significant changes are observed in secondary structure of p20 gene (Fig. 4).
The results also define that the genotypes identical to Pakistani isolates genotypes are spread to quite distant places around the world. However, not much divergence has been observed in the secondary sequence of p20 protein of different genotypes. The isolates that spread indigenously in Pakistan are closely related to each other genotypically and there is no divergent infection. A similar result was observed in Spain, California and Italy in various studies, concluding that mixed infections rarely occur and are transient [29-31]. Therefore, it is inferred that genetically similar isolates have been introduced in Pakistan in various events. Within the country CTV is widely spread in Punjab and is commonly found in Mosambi sweet orange. Iftikhkar et al., reported that the disease is found in four districts of Punjab namely, Sahiwal, Sargodha, Sheikhupura and Faisalabad [32]. The infection frequency varied in these areas with the highest rate recorded to be 18.18% in Sahiwal and lowest 7.14% in Sheikhupura. The virus is assumed to be spread mainly due to grafting [15, 33]. Infected trees also serve as reservoirs for the virus. The virus spread in KPK and northern areas of Pakistan is also due to aphid vectors [34] of which A. gossiypii is most efficient. It has been reported that a single A. gossiypii aphid can successfully transmit CTV from sour orange to sweet orange and back from sweet orange to sour orange [35]. Biogeography of the CTV isolates, used in this study (Fig. 5), concludes that how this pathogenic virus diffused in Pakistan in connections with foreign isolates. If in any of these countries an Integrated Disease Management (IDM) system exists with notable success rate can be adopted to prevent further economic losses. Strategies such as Quarantine and budwood certification programs can be introduced. In the endemic countries CTV tolerant root stock or introduction of genetically engineered resistance in the plants are some potential ways to avoid the infection [36].
There are several reports present worldwide accounting for slow evolution rate of Citrus tristeza virus and its consistently conserved sequences in the 3' region of the genome. Various regions such as 3' and 5' end, ORF, CP and CPm of the genome has been utilized to differentiate different isolates of CTV [37]. Solinska et al., reported RNA recombination as being the chief mechanism in plus strand virus evolution [38]. RNA recombination not only produces genetic variability in viruses; it also has a genome repair mechanism to fix damaged RNA [39]. Nevertheless, for CTV, RNA recombination is strongly involved in its evolution [37, 40]. Comparison of p20 gene showed slow divergence of virus in various regions of the world and no actual divergence is present among Pakistani isolates and that of Egypt, China, India, Italy, Argentina and South Africa. Moreover, the secondary structure of protein is slightly mutated among isolates dispersed in geographically distant places. However, mutations at certain points may affect the activity of the protein as the amino acids replaced by other amino acids have distinct properties i.e. hydrophobic amino acids have been replaced by polar ones and positively charged amino acids have been altered with negatively charged amino acids. Most of amino acid substitution has been observed at the 5' end in Pakistani isolates seldom nucleotide mutations affected the middle amino acid sequence of the protein. It is deduced that Pakistani isolates have a similar pattern of variability in their nucleotides and this pattern is not very distinct from the other isolates worldwide.
Our study has confirmed multiple introductions of CTV in Pakistan and has also confirmed dissemination of CTV within Pakistan. The p20 gene is involved in suppression of RNA silencing induced by host plant and also is the main component of amorphous inclusion bodies formed in citrus plants. The present study shows that the conserved and variable regions of the gene are involved in suppression of plant’s immune response and provides valuable data for designing artificial microRNAs against CTV. This is one of the first studies conducted with reference to evolution and emergence of CTV in Pakistan. Very few data is available related to CTV prevalence in Pakistan and many regions of the CTV genome needs to be sequenced in order to perform phylogeographical and phylodynamic study of the virus. This study provides insight into spread of CTV virus in Pakistani citrus varieties. This study also shows that the mutations are present in the predicted secondary structure of the p20 protein. However, it is not known whether it affects the pathogenicity of the virus; due to lack of tertiary structure. There is a need to construct tertiary structure for p20 in order to further investigate its role in pathogenicity and its relevant control.
This study gives significant insights into Citrus tristeza virus spread and diversity in Pakistan on the basis of p20 gene sequence variability in comparison to worldwide isolates. On the basis of this scientific revision we recommend scientists that in future encoders wet lab researcher of Citrus tristeza virus in Pakistan should carry out whole genome sequencing of this economically important pathogenic virus. As bioinformatics procedure, in molecular phylogenetic context, are well elaborative now days [23]. Therefore, a whole genome phylogeny may lead us towards a more comprehensive understanding of the virus diversity and spread.
References
- Talon M, Gmitter Jr FG. Citrus genomics. International Journal of Plant Genomics, (2008); 2008: 1-17.
- Atta S, Zhou C, Zhou Y, CAO M, Wang X. Distribution and research advances of Citrus tristeza virus. Journal of Integrative Agriculture, (2012); 11: 346-358.
- Agriculture Statistics of Pakistan, 2011. Agriculture Statistics of Pakistan. Government of Pakistan Statistics Division Pakistan Bureau of Statistics, Islamabad, (2011): p. 89.
- Febres VJ, Ashoulin L, Mawassi M, Frank A, Bar-Joseph M, et al. The P27 protein is present at one end of citrus tristeza virus particles. Phytopathology, (1996); 86: 1331–1335.
- Biswas KK, Tarafdar A, Sharma SK. Complete genome of mandarin decline Citrus tristeza virus of Northeastern Himalayan hill region of India: comparative analyses determine recombinant. Archives of Virology, (2012b); 157: 579-583.
- Dolja VV, Kreuze JF, Valkonen JP. Comparative and functional genomics of Closteroviruses. Virus Research (2006); 117: 38–51.
- Satyanayanana T, Gowda S, Mawassi M, Albiach-Martí MR, Ayllón MA, et al. Closterovirus encoded HSP70 homolog & p61 in addition to both coat proteins function in efficient virion assembly. Virology, (2000); 278: 253-265.
- Tatineni S, Robertson CJ, Garnsey SM, Bar-Joseph M, Gowda S, et al. Three genes of Citrus tristeza virus are dispensable for infection and movement throughout some varieties of citrus trees. Virology, (2008); 376(2): 297-307.
- Lu R, Folimonov A, Shintaku M., Li WX, Falk B W, et al. Three distinct suppressors of RNA silencing encoded by a 20-kb viral RNA genome. Proceedings of the National Academy of Sciences of the United States of America, (2004); 101: 15742–15747.
- Voinnet O. Induction and suppression of RNA silencing: insights from viral infections. Nature Review of Genetics, (2005); 6: 206-220.
- Iftikhar Y, Shahid M, Khan M A, Naqvi S A. Protein analysis of stem bark from three citrus varieties infected with citrus tristeza virus in Pakistan, (2012); 24(2): 167-169.
- Ayazpour K, Sijam K, Vadamalai G, Jafar H. Status of Citrus Tristeza Virus (CTV) in Peninsular Malaysia. African Journal of Microbiology Research, (2011); 5: 838-843.
- Silva G, Marques N, Nolasco G. The evolutionary rate of Citrus tristeza virus ranks amongst the rates of slowest RNA viruses. Journal of General Virology, (2012); 93: 419-429.
- Moreno P, Ambro´s S, Albiach-Martı´ MR, Guerri J, Pen˜ a L. Citrus tristeza virus: A pathogen that changed the course of the citrus industry. Molecular Plant Pathology, (2008); 9: 251–268.
- Catara A, Azzaro A, Davino M, Grimaldi V, Hussain M, Saleem M, Mirza M S 1991. A survey of tristeza and greening in Punjab, Pakistan. In Proceedings of 11th Conference of the International Organization of Citrus Virologist. Riverside, University of California: 166-170.
- Melzer MJ, Borth WB, Sether DM, Ferreira S, Gonsalves D, et al. Genetic diversity and evidence for recent modular recombination in Hawaiian Citrus tristeza virus. Virus Genes, (2010); 40: 111-118.
- Roy A, Ananthakrishnan G, Hartung JS, Brlansky RH. Development and application of a multiplex reverse-transcription polymerase chain reaction assay for screening a global collection of Citrus tristeza virusisolates. Phytopathology (2010); 100: 1077-1088.
- Hilf ME, Mavrodieva VA, Garnsey SM. Genetic marker analysis of a global collection of isolates of Citrus tristeza virus: Characterization & distribution of CTV genotypes & association with symptoms. Phytopathology, (2005); 95: 909-917.
- López C, Ayllón MA, Navas-Castillo J, Guerri J, Moreno P, et al. Molecular variability of the 5' & 3' terminal regions of citrus tristeza virus RNA. Phytopathology, (1998); 88: 685-691.
- Vives MC, Rubio L, López C, Navas-Castillo J, Albiach-Martí MR, et al. The complete genome sequence of the major component of a mild citrus tristeza virus isolate. Journal of General Virology, (1999); 80: 811-816.
- Harper SJ. Citrus tristeza virus: evolution of complex and varied genotypic groups. Frontiers of Microbiology, (2014): 77.
- Scott K A, Hlela Q, Zablocki O, Read D, van Vuuren S, Pietersen G. Genotype composition of populations of grapefruit-cross-protecting citrus tristeza virus strain GFMS12 in different host plants and aphid transmitted sub-isolates. Archives of Virology, (2013); 158: 27- 37.
- Yang Z, Rannala B. Molecular phylogenetics: principles and practice. Nature Reviews Genetics, (2012); 13: 303–314.
- Nasir A, Caetano-Anollés G. A phylogenomic data-driven exploration of viral origins and evolution. Science Advances, (2015); 1:8, e1500527.
- Darriba D, Taboada GL, Doallo R, Posada D. "jModelTest 2: more models, new heuristics and parallel computing". Nature Methods (2012); 9: 772.
- Huelsenbeck, Ronquist. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics, (2001); 17: 754-755.
- Albiach-Martı´ MR, Mawassi M, Gowda S, Satyanarayana T, Hilf ME, et al. Sequences of Citrus tristeza virus separated in time and space are essentially identical. Journal of Virology, (2000); 74: 6856–6865.
- Rubio L, Ayllo´ n MA, Kong P, Fernandez A, Polek ML, et al. Genetic variation of Citrus tristeza virus isolates from California and Spain: Evidence for mixed infections and recombination. Journal of Virology, (2001); 75: 8054–8062.
- Davino S, Willemsen A, Panno S, Davino M, Catara A, et al. Emergence and phylodynamics of Citrus tristeza virus in Sicily, Italy. PLOS One, (2013); 8: e66700.
- d’Urso F, Sambade A, Moya A, Guerri J, Moreno P . Variation of haplotype distributions of two genomic regions of Citrus tristeza virus populations from Eastern Spain. Molecular Ecology, (2003); 12: 517–526.
- Kong P, Rubio L, Polek ML, Falk BW. Population structure and genetic diversity within California Citrus tristeza virus (CTV) isolates. Virus Genes, (2000); 21: 139–145.
- Iftikhar Y, Aslam K, Rashid A, Mughal S M, Iqbal Z, Batool A, Abbas M, Khan M M, Muhammad S, Jaskani M J. Occurrence and distribution of Citrus Tristeza Closterovirus in the Punjab and NWFP, Pakistan. Pakistan Journal of Botany, (2009); 41(1): 373-380.
- Atta S, Liu Y, Cao M, Yang F Y, Zhou Y., et al. Molecular characterization of Citrus tristeza virus isolates from Pakistan based on CPG/Hinf I restriction fragment length polymorphism (RFLP) groups analysis. African Journal of Biotechnology, (2013) 10(44): 8689-8694.
- Arif M, Khan W, Ibrahim M, Fahim, M. Citrus tristeza virus: An increasing trend in the virus occurrence and distribution in citrus fruits of Northwest, Pakistan. African Journal of Biotechnology, (2005); 14(30): 2352-2360.
- Arif M, Ahmad A, Ibrahim M, Hassan S. Occurrence and distribution of virus and virus-like diseases of citrus in north-west frontier province of Pakistan. Pakistan Journal of Botany, (2005); 37(2): 407.
- Atta S, Zhou C Y, Yan Z, Cao M J, Wang X F. Distribution and research advances of Citrus tristeza virus. Journal of Integrative Agriculture. (2012); 11(3): 346-358.
- Xiao-yun W, Xiao-fei C, Lu L, Xiao-xia W. Genetic diversity and global distribution of Citrus tristeza virus (CTV) strains. Journal of Northeast Agricultural University, (2012); 19: 9-18.
- Sztuba-Solinska J, Urbanowicz A, Figlerowicz M, Bujarski JJ. RNA RNA recombination in plant virus replication and evolution. Annual Review of Phytopathology, (2012); 49: 415-443.
- Barr J N, Fearns R. How RNA viruses maintain their genome integrity. Journal of General Virology, (2010); 91: 1373-1387.
- Martin S, Sambade A, Rubio L, Vives MC, Moya P, et al. Contribution of recombination and selection to molecular evolution of Citrus tristeza virus. Journal of General Virology, (2009); 90: 1527-1538.