

Full Length Research Article
Virtual Screening of Compounds for the Identification of Potential Drug Candidates Targeting the RACK1 Receptor in Liver Cancer
Bandar Hamad Aloufi*
Adv. life sci., vol. 12, no. 1, pp. 237-244, February 2025
*– Corresponding Author: Bandar Hamad Aloufi (Email: Bandaraloufi@yahoo.com)
Authors' Affiliations
[Date Received: 08/10/2024; Date Revised: 27/12/2024; Date Published: 31/12/2024]
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related deaths globally and the sixth most common cancer, particularly in the Asia-Pacific and African regions. Liver cirrhosis, a critical precursor to HCC and liver failure, necessitates effective treatment options. Although surgical intervention is the current standard, there is a pressing need for novel therapeutics with improved efficacy and reduced side effects. This study focuses on RACK1 (Receptor for Activated C-Kinase 1), a pivotal protein in cancer progression, as a therapeutic target for HCC.
Methods: Protein structures of overexpressed genes in HCC, including RACK1, were retrieved from the Protein Data Bank (PDB). Active binding sites on RACK1 were identified for potential ligand interactions. A library of 12,000 phytochemicals was sourced from PubChem, ZINC, and MP3D databases and screened against RACK1 using the PyRx virtual screening tool. The top candidates were analyzed for pharmacokinetic properties using ADMETsar. Molecular dynamics simulations were conducted to study ligand-receptor interactions and validate the potential drug candidates.
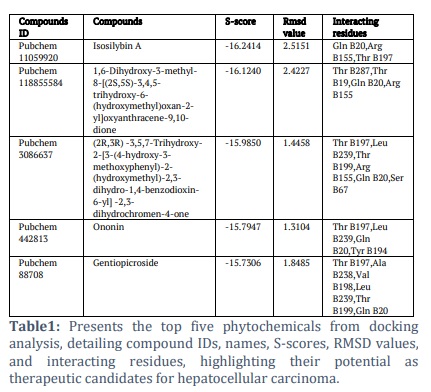
Results: The study identified promising phytochemical compounds (Pubchem11059920, Pubchem118855584, Pubchem3086637, Pubchem442813 and Pubchem88708) capable of binding to RACK1 with high affinity. These compounds exhibited favorable ADMET properties, indicating their potential as drug candidates. Molecular dynamics simulations confirmed stable and significant interactions between the identified ligands and RACK1, supporting their inhibitory potential.
Conclusion: This research highlights RACK1 as a viable therapeutic target for HCC. The identified drug candidates demonstrate potential to inhibit RACK1 function, offering a pathway to suppress HCC progression at its early stages. These findings provide a foundation for the development of effective and targeted treatments for HCC.
Keywords: Hepatocellular carcinoma (HCC), RACK1 receptor, Phytochemical screening, Virtual screening, Molecular dynamics simulation
Introduction![]()
Liver cancer is the leading cause of cancer death worldwide, and it is the fifth leading cause of cancer death in the United States of America [1]. It is also the only one of the top five most lethal cancers that have seen an increase in the number of persons who develop it on a yearly percentage basis [2]. Liver disorders are more common in developing countries [3]. Hepatitis B and C viruses, fatty liver disease, alcohol-related cirrhosis, smoking, obesity, diabetes, iron overload, and other dietary exposures are all risk factors. Liver cancer has a dismal prognosis.
The early signs of liver cancer are difficult to detect [4]. The majority of patients is diagnosed at an advanced stage and misses out on the best surgical options [5].RACK1 is a tryptophan–aspartate repeat (WD repeat) protein [6]. It's a standard scaffold protein for several kinases and receptors, and it's engaged in a wide range of biological responses [7]. In a previous investigation, RACK1 was discovered to be present in both ribosome-bound and non-ribosomal-bound versions of the 40S ribosome subunit [7].
According to previous research, RACK1 is a 40S ribosomal subunit component that persists across both bound and unbound forms [8]. RACK1 (receptor for activated C kinase 1) was discovered because of its capacity to attach the activated form of protein kinase C, (PKC). Trp-Asp (WD) repeat proteins have been identified as an adaptor protein involved in several intracellular signaling pathways. RACK1 mRNA and protein levels are higher in clinical samples from patients with hepatocellular carcinoma [9,10].
RACK1 expression is linked to both the clinical stage and the poor prognosis of cancer patients [10]. By binding to and activating the JNK specific upstream kinase, RACK1 overexpression enhances HCC growth by increasing JNK activity MKK7 [10]. Furthermore, ribosomal RACK1 works with PKCII to phosphorylate eIF4E, leading to preferential translation of growth and survival factors [11].
In current study drug candidates against the protein RACK1 was identified. Phytochemicals were docked with the RACK1 protein's catalytic triad to test their suitability as inhibitors. After docking, almost 1000 compounds of RACK1 protein with top conformations from 12000 complexes were selected. Out of 1000 best compounds five best complexes were finalized on the basis of minimum S score, RMSD value and having good interactions. Insilico approach of Molecular simulation is also used to pre-screen the virtual compounds database [12].
Five compounds—Isosilybin A, 1,6-Dihydroxy-3-methyl-8-[(2S,5S)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyanthracene-9,10-dione, (2R,3R)-3,5,7-Trihydroxy-2-[3-(4-hydroxy-3-methoxyphenyl)-2-(hydroxymethyl)-2,3-dihydro-1,4-benzodioxin-6-yl]-2,3-dihydrochromen-4-one, Ononin, and Gentiopicroside—exhibited key characteristics required for potential drug candidates. Additionally, the pharmacokinetic profiles of these compounds were analyzed through ADMETsar using the SwissADME server.
Recent research revealed that these molecules displayed the highest docking scores, indicating strong and stable interactions with the overexpressed RACK1 protein. These findings could contribute to the identification of promising therapeutic targets for RACK1.
Methods![]()
Structure Retrieval
The RACK1 protein crystal structure was obtained from the Protein Data Bank with PDB ID:4AOW [13] Preparation was done with parameters MMFF94X+Solvation, force field, and 3D protonation was used to remove water molecules, minimize energy, and execute 3D protonation. Chiral, 0.05 gradient Current geometry is a constraint [14].
Preparation of ligand library
An extensive survey was conducted in order to identify phytochemicals that are effective and beneficial in the treatment of liver cancer disease. 5000 phytochemicals were retrieved from medicinal plant databases like MPD3 [15], CHEMBL [16], PubChem [17] and ZINC [18] for molecular docking ready to dock library was prepared.
After minimizing the energy with the following parameters: gradient: 0.05, Force Field: MMFF94X, Chiral Constraint: Current Geometry, Every ligand molecule was saved in.pdb format in the PyRx. Molecular docking has been done against the RACK1 protein using the PyRx tools in order to conduct an inhibitor scan [14].
Molecular Docking
Molecular docking is method for determining the binding orientation of tiny molecules to their targets. Thus, a technique in drug discovery and screening for new compounds against these horrible and difficult diseases is critical. Receptor was obtained from PDB in 3D crystal structure [13]. 3D protonation and Energy minimization had used to refine the structure. Molecular docking is an important tool for computer aided drug designing. Docking is an approach which forecasts the binding mode of a ligand with protein whose 3D structural is known.
It basically checks the interaction between two compounds. The ready to dock library of 12000 phytochemicals was docked with the key interacting residues of the RACK1 protein structure. Different conformations of ligand were obtained through PyRx. It was determined that the parameters for docking were 1 for rescoring, 2 for London dG, and 10 for refinement.
Ligand receptor interaction analysis
Large library of compounds is virtually screened and results are checked. The receptor-ligand interactions of complexes were analyzed using the LigX tool [19]. It gives the good picture of receptor-ligand interactions of best docked complexes. 2D plots of receptor-ligand interactions were evaluated.
It shows the hydrogen bonding, hydrophobic interactions, electrostatic interactions, and van der wall forces accountable for drug like molecule's affinity in actively-docked pockets. ChimeraX was used to generate 3D images of the protein inhibitor complexes.
Physiochemical property profile
Drug likeness properties of best dock complexes were studied using molinspiration server (https://www.molinspiration.com). This server provides prediction based “Lipinski rule of five”. The Lipinski rule of five includes molecular characteristics parameters such as a logP value of less than five and a MW of fewer than 500 Daltons. While the hydrogen bond donor must be less than five and the hydrogen bond acceptor must be less than ten, the hydrogen bond acceptor must be less than five.
ADMET properties were accessed using swissADME (http://www.swissadme.ch/) Absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis was conducted [20].
Molecular Dynamics Simulation Protocol
The stability of the docked complexes was analyzed using a 200 ns Molecular Dynamics simulation research experiment. Schrodinger's Desmond Simulation Package was used to examine the complex in an OPLS3 force field. Maintain system temperature (300 K) and pressure (1 bar).
Following 1000 steps of steepest descent, conjugate gradient methods were used to reduce energy consumption.
Results![]()
Retrieval of structural and ligand libraries
Using the Protein Data Bank (PDB), we determine the three-dimensional structure of RACK1. The high resolution of this receptor made it an excellent choice as a target. Its resolution was 2.45Å.
The 3D structure of RACK1 shown in the Supplementary File 1, Figure S1. Structures was improved and then employed as receptor. Docking was carried out utilizing ligands from the PubChem, ZINC, and MPD3 libraries, which contained 12000 phytochemicals in total.
Molecular docking
This section presents the comprehensive results obtained from docking the receptor protein structures with a diverse library of phytochemicals. Each compound generated ten distinct conformations, providing a wide range of binding possibilities. These conformations were carefully evaluated and ranked according to several key parameters, including S scoring, root-mean-square deviation (RMD) values, and the nature of their interactions with the active sites of the target proteins.
The scoring metrics allowed for the identification of the most promising candidates, highlighting their ability to form stable and strong bonds with the receptor's active pockets. After a detailed assessment, the top five compounds were selected based on their lowest docking scores, which reflect stronger binding affinities and more stable interactions with the receptor protein.
These leading candidates were then prioritized for further analysis, including pharmacokinetic evaluations and molecular dynamics simulations. As shown in Supplementary file 1, Figure S2, compounds exhibited superior binding characteristics, making them potential candidates for future drug development targeting the specific receptor protein.
The selected compounds exhibited significant interactions with the binding pockets of the target proteins, forming stable complexes that suggest strong affinity. These interactions were reflected in their exceptionally low binding energy values, indicating favorable binding efficiency.
Among all docked ligands, these compounds consistently ranked with the lowest binding energies, as detailed in Table 1. This highlights their potential as promising candidates for further therapeutic development.
Receptor-Ligand interaction
The interactions between the receptor protein and the top five selected compounds were thoroughly analyzed to assess their binding efficiency. Using the Lig X tool, 2D visualizations of the receptor-ligand interactions were generated for the best-docked complexes. Upon docking the RACK1 protein with a comprehensive library of 12,000 phytochemicals, Isosilybin A emerged as the most promising candidate.
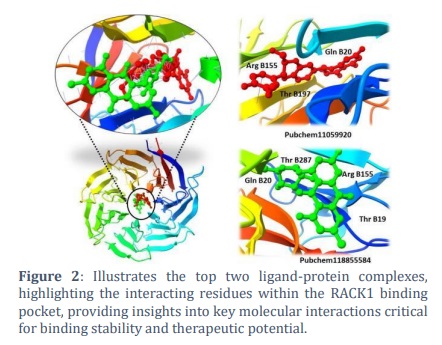
This compound demonstrated strong binding affinity, with a docking score of -16.24 kcal/mol, and formed stable interactions with key amino acid residues, including Gln B20, Arg B155, and Thr B197. Its superior binding characteristics earned it the highest rank among all screened ligands. Following PubChem11059920, PubChem118855584 was ranked as the second most effective compound.
It exhibited notable interactions with Thr B287, Thr B19, Gln B20, and Arg B155, with a slightly lower binding energy score of -16.12 kcal/mol. These results, illustrated in Figure 2, highlight the robust binding potential of both compounds, positioning them as strong candidates for further exploration in drug development targeting RACK1. The favorable docking scores and interactions with critical residues suggest these compounds could play a significant role in inhibiting the function of the RACK1 protein, making them ideal for subsequent pharmacological studies.
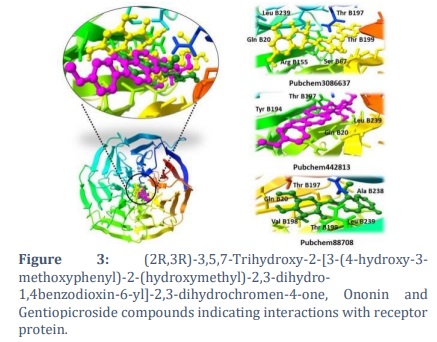
Moreover, (2R,3R)-3,5,7-Trihydroxy-2-[3-(4-hydroxy-3-methoxyphenyl)-2-(hydroxymethyl)-2,3-dihydro-1,4benzodioxin-6-yl]-2,3-dihydrochromen-4-one, Ononin and Gentiopicroside demonstrated the binding affinity of -15.98 kcal mol-1, -15.79 kcal mol-1 and-15.73 kcal mol-1 and interaction with the sites Thr B197,Leu B239,Thr B199,Arg B155,Gln B20,Ser B67, Thr B197,Leu B239,Gln B20,Tyr B194, Thr B197,Ala B238,Val B198,Leu B239,Thr B199,Gln B20 as shown in the Figure 3.
Druglikeness Prediction
To evaluate the drug-like properties of the most promising ligands, the Lipinski's Rule of Five (RO5) was applied to computationally screen their physicochemical characteristics. According to RO5, a compound qualifies as drug-like if it has a molecular weight of ≤ 500 g/mol, contains no more than 5 hydrogen bond donors and 10 hydrogen bond acceptors, and has a miLog P value of less than 5. A potential drug candidate may still be considered acceptable even if it violates one of these criteria.
As shown in (Supplementary file 1, Table S1), the top phytochemical hits, along with the reference compound, were analyzed for their predicted drug-likeness. All the selected ligands demonstrated favorable drug-like properties, meeting most of the criteria, and showing strong potential for further development.
ADMET Profiling
ADME Pharmacokinetic parameters were assessed using ADME and AdmetSAR tools to thoroughly evaluate the absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles of the top therapeutic candidate compounds. These pharmacokinetic factors are crucial for determining the suitability of a compound for drug development.
The ADMET properties of the derived phytochemicals targeting both receptors are presented in (Supplementary file 1, Table S2). Many potential drugs fail to advance through the development pipeline due to poor pharmacokinetic profiles or toxic effects. Thus, early-stage drug discovery heavily relies on high-throughput and efficient ADMET screening to filter out unsuitable candidates and identify promising lead compounds. In this study, ADMET profiling revealed that none of the selected compounds exhibited negative effects on absorption, a critical parameter for drug viability.
Additionally, the overall pharmacokinetic and toxicity results were favorable, with no significant safety concerns identified.
These findings suggest that the candidate compounds possess excellent ADMET characteristics, indicating their potential as viable therapeutic candidates for further preclinical development. By demonstrating both effective pharmacokinetics and minimal toxicity, these compounds stand out as promising leads for future drug design and optimization efforts.
MD Simulation
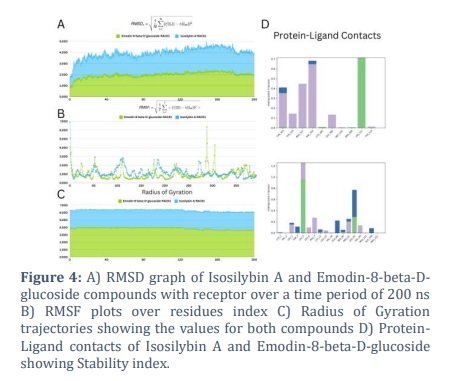
For a better understanding of the molecular insights that are involved in the binding of Isosilybin A and Emodin-8-beta-D-glucoside in the active pocket of the 4AOW protein, a 200-ns molecular dynamic simulation was run.
Measuring the root mean squared deviations (RMSD) of the docking complex Isosilybin A and Emodin-8-beta-D-glucoside during the production run allowed us to determine the stabilization of the complex. The root mean square deviation (RMSD) for the complex Isosilybin A revealed a modest fluctuation of approximately 1.0 and a consistent trajectory during the production run, with a maximum stability shown in Figure 4A.
The RMSF analysis of the complex Isosilybin A showed RMSF values between 1.3 and 1.6 with local ligand-contact fluctuation at several points, with extremes at residues 280 for both chains, as shown in Figure 4B. RoG analysis was done to check the stability of both compounds with receptor as shown in the Figure 4C. Figure 4D shows different kinds of protein-ligand contacts.
Figures & Tables
Liver cancer has rapidly become a significant global health concern, claiming millions of lives each year. The increasing prevalence of this disease has placed a heavy burden on healthcare systems worldwide, making the discovery of effective treatments a pressing issue. Through extensive literature surveys and research, specific upregulated genes associated with liver cancer have been identified, offering potential molecular targets for therapeutic intervention. The quest to find effective treatments for liver cancer has traditionally been a laborious and time-consuming process. Conventional drug design methods, which involve years of experimentation and clinical trials, often take a long time to develop novel drugs. This lengthy process, while thorough, delays the availability of potentially life-saving treatments for patients in urgent need. In recent years, the rise of computer-based approaches has revolutionized drug discovery and design. These in silico techniques have provided researchers with powerful tools to predict and analyze the interactions between potential drug candidates and their molecular targets before physical testing or manufacturing in the laboratory. This computational shift has significantly reduced the time and costs associated with traditional drug development. By virtually simulating the binding of compounds to target proteins, researchers can rapidly screen thousands of compounds, narrowing down the most promising candidates for further investigation. This method accelerates the entire drug development pipeline, allowing researchers to focus on compounds with the highest likelihood of success in combating diseases like liver cancer. The traditional process of drug design requires a long time not only to identify effective compounds but also to conduct extensive preclinical and clinical testing to ensure safety and efficacy. However, the introduction of computational techniques in drug discovery has dramatically shortened this timeline [21]. Computer-based drug design, or in silico drug development, allows researchers to test vast libraries of compounds for their potential as effective drugs in a fraction of the time needed for conventional approaches. This method has proven particularly valuable in reducing the risk of failure in later stages of development, as compounds are already vetted for key properties such as binding affinity, selectivity, and pharmacokinetic behavior before entering physical trials [22]. The ability to predict a compound's drug-likeness early in the process helps minimize unnecessary costs and resources associated with testing compounds that are unlikely to succeed [23]. Moreover, in silico approaches offer more than just efficiency in terms of time; they are also cost-effective [24]. Traditional drug discovery often involves high costs due to the need for labor-intensive laboratory experiments, expensive materials, and prolonged trial phases [25]. By contrast, computational methods eliminate many of these barriers. Researchers can use sophisticated algorithms to simulate molecular interactions and predict how compounds will behave in the body without the need for expensive reagents or extensive laboratory work. As a result, the overall financial burden of drug discovery is significantly reduced, making it possible to explore a wider range of potential treatments at a lower cost. With the rapid advancements in bioinformatics, new tools and algorithms have emerged that enable researchers to precisely identify and target disease-causing molecules. These cutting-edge techniques have transformed the way scientists approach drug discovery, making it possible to discover new therapeutic compounds with unprecedented speed and accuracy. One of the most widely used in silico techniques is molecular docking, which has become an invaluable tool in modern medical research. Molecular docking involves predicting the orientation of small molecules as they bind to target proteins, allowing researchers to determine which compounds have the most favorable interactions with the disease-causing proteins. By understanding how small molecules interact with their targets, researchers can design drugs that more effectively combat diseases such as liver cancer. Molecular docking has played a critical role in this study, as it allows scientists to explore how various compounds bind to the active sites of liver cancer-related proteins. By simulating the interactions between these compounds and their protein targets, molecular docking provides valuable insights into which molecules are most likely to succeed as drugs. This method enables researchers to predict the binding affinity of a compound to its target protein, identify favorable interactions with key residues, and assess the overall stability of the drug-protein complex. As a result, molecular docking has become a central approach for identifying new compounds that may inhibit the function of upregulated proteins in liver cancer, potentially leading to the discovery of highly effective drugs. The growing reliance on molecular docking and other in silico methods underscores the tremendous potential these techniques have in revolutionizing the field of drug discovery[26]. By harnessing the power of computational tools, researchers can identify drug candidates that not only show strong binding interactions but also possess desirable drug-like properties. This approach paves the way for the development of more targeted and effective therapies, particularly for complex diseases like liver cancer where time is of the essence [27]. The advancements in bioinformatics have made it possible to develop drugs faster, more efficiently, and with fewer side effects, ultimately providing new hope for millions of people suffering from liver cancer around the world [28].
This study has demonstrated that by pinpointing the active sites of target proteins, we can potentially suppress their activity and reduce disease progression. Specifically, several compounds exhibited strong interactions with the RACK1 protein, which is known to be overexpressed in certain disease conditions, such as cancer[29]. The molecular characteristics and drug-likeness of the selected compounds were thoroughly assessed using the Lipinski Rule of Five[30]. According to this rule, an ideal drug candidate should have a molecular weight of less than 500 Daltons, no more than five hydrogen bond donors, fewer than 10 hydrogen bond acceptors, and an AlogP value (a measure of lipophilicity) below five[31]. These criteria help in predicting a compound’s ability to be absorbed and function effectively within the human body.
The compounds identified in this study adhered to all of Lipinski’s parameters, showing no violations of the rule, which is a critical indicator of their potential as drug candidates. Additionally, these compounds had low docking scores, suggesting strong binding affinity with the protein, and root-mean-square deviation (RMSD) values below 3, indicating the reliability of the docking process [32,33]. Such low RMSD values reinforce the stability and accuracy of the predicted ligand-protein interactions. However, fulfilling the Lipinski Rule of Five is just the beginning; ensuring that a compound has suitable pharmacokinetic properties is equally important. ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) analysis is a crucial, yet complex, stage in drug discovery, as it helps to predict how a compound behaves inside the body. This was accomplished using the SwissADME database, a reliable resource for assessing the pharmacokinetic profile of compounds [34,35]. The results of the ADMET analysis revealed that the selected compounds exhibited favorable pharmacokinetic properties, meaning they are likely to be well-absorbed, adequately distributed throughout the body, efficiently metabolized, and exhibit minimal toxicity. These characteristics are essential for any compound being considered for therapeutic use [36,37]. Based on the molecular docking and pharmacokinetic evaluations, it can be concluded that the identified phytochemicals have the potential to inhibit the activity of RACK1 by interacting with its binding pockets. By effectively targeting this protein, these compounds could disrupt the pathways that contribute to disease progression. Therefore, these phytochemicals represent promising drug candidates for the development of new therapeutic strategies, particularly against conditions where RACK1 is implicated, such as cancer. Further experimental validation, including in vitro and in vivo studies, will be essential to confirm their efficacy and potential for clinical use.
The primary aim of this study is to identify novel compounds that could serve as effective drug candidates targeting the highly overexpressed RACK1 gene. Advances in bioinformatics have introduced a range of tools and algorithms that significantly contribute to the identification of drug targets. PyRx, a key tool in the molecular docking process, was utilized for virtual screening in this drug discovery effort. The receptor proteins were docked with a pre-compiled library of compounds, and the resulting interactions were thoroughly evaluated based on various parameters, including Lipinski's Rule of Five and ADMET analysis. ADMET profiling assessed the pharmacokinetics of the compounds, examining their absorption in the intestine, distribution properties, ability to cross the blood-brain barrier, metabolism, and potential toxicity. The results revealed that the selected compounds exhibited favorable drug-like properties, aligning with essential criteria for effective drug candidates. This study's findings suggest that these compounds possess significant potential as therapeutic agents, specifically by inhibiting the activity of overexpressed proteins involved in dysregulated gene expression. Further experimental validation of these phytochemicals is highly recommended in future research to confirm their efficacy and therapeutic potential.
Supplementary Data
Readers may contact the corresponding author for supplementary data/information.
The author declare that there is no conflict of interest regarding the publication of this paper.
![]() References
References
- Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology, (2010); 51(5): 1820-1832.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: a cancer journal for clinicians, (2018); 68(1): 7-30.
- Center MM, Jemal A. International trends in liver cancer incidence rates. Cancer Epidemiology and Prevention Biomarkers, (2011); 20(11): 2362-2368.
- Bosch FX, Ribes J, Díaz M, Cléries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology, (2004); 127(5): S5-S16.
- Liu C-Y, Chen K-F, Chen P-J. Treatment of liver cancer. Cold Spring Harbor perspectives in medicine, (2015); 5(9): a021535.
- McCahill A, Warwicker J, Bolger GB, Houslay MD, Yarwood SJ. The RACK1 scaffold protein: a dynamic cog in cell response mechanisms. Molecular pharmacology, (2002); 62(6): 1261-1273.
- Adams DR, Ron D, Kiely PA. RACK1, A multifaceted scaffolding protein: Structure and function. Cell communication and signaling, (2011); 9(1): 1-24.
- Li J, Xie D. RACK1, a versatile hub in cancer. Oncogene, (2015); 34(15): 1890-1898.
- Yoon SY, Kim J-M, Oh J-H, Jeon Y-J, Lee D-S, et al. Gene expression profiling of human HBV-and/or HCV-associated hepatocellular carcinoma cells using expressed sequence tags. International journal of oncology, (2006); 29(2): 315-327.
- Guo Y, Wang W, Wang J, Feng J, Wang Q, et al. Receptor for activated C kinase 1 promotes hepatocellular carcinoma growth by enhancing mitogen‐activated protein kinase kinase 7 activity. Hepatology, (2013); 57(1): 140-151.
- Ruan Y, Sun L, Hao Y, Wang L, Xu J, et al. Ribosomal RACK1 promotes chemoresistance and growth in human hepatocellular carcinoma. The Journal of clinical investigation, (2012); 122(7): 2554-2566.
- Halim SA, Sikandari AG, Khan A, Wadood A, Fatmi MQ, et al. Structure-based virtual screening of tumor necrosis factor-α inhibitors by cheminformatics approaches and bio-molecular simulation. Biomolecules, (2021); 11(2): 329.
- Burley SK, Bhikadiya C, Bi C, Bittrich S, Chen L, et al. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic acids research, (2021); 49(D1): D437-D451.
- Rehman G, Khan S., Iqbal A, Hamayun M, Hussain A, et al. Confirmation of antioxidant and antiangiogenic potential of adult housefly extracts through chick chorioallantoic membrane (cam) assay. FEB FRESENIUS ENVIRONMENTAL BULLETIN,(2020); p.8620.
- Muneer I, Ahmad S, Naz A, Abbasi SW, Alblihy A, et al. Discovery of Novel Inhibitors From Medicinal Plants for V-Domain Ig Suppressor of T-Cell Activation. Frontiers in Molecular Biosciences, (2021); 8, p.716735.
- Said MA, Albohy A, Abdelrahman MA, Ibrahim HS. Importance of glutamine 189 flexibility in SARS-CoV-2 main protease: Lesson learned from in silico virtual screening of ChEMBL database and molecular dynamics. European Journal of Pharmaceutical Sciences, (2021); 160105744.
- Kim S, Chen J, Cheng T, Gindulyte A, He J, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic acids research, (2021); 49(D1): D1388-D1395.
- Berge A. and Björck L. Streptococcal Cysteine Proteinase Releases Biologically Active Fragments of Streptococcal Surface Proteins. Journal of Biological Chemistry, (1995); 270(17), pp.9862-9867.
- Rehman A, Ashfaq UA, Javed MR, Shahid F, Noor F, et al. The Screening of phytochemicals against NS5 Polymerase to treat Zika Virus infection: Integrated computational based approach. Combinatorial Chemistry & High Throughput Screening, (2022); 25(4), pp.738-751.
- Riyadi P, Sari I, Kurniasih R, Agustini T, Swastawati F, et al. SwissADME predictions of pharmacokinetics and drug-likeness properties of small molecules present in Spirulina platensis; 2021. IOP Publishing. pp. 012021.
- Snowden FM. Emerging and reemerging diseases: a historical perspective. Immunological reviews, (2008); 225(1): 9-26.
- Water S, Organization WH Emerging issues in water and infectious disease. 2002; World Health Organization.
- Schneider G, Fechner U. Computer-based de novo design of drug-like molecules. Nature reviews Drug discovery, (2005); 4(8): 649-663.
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature reviews Drug discovery, (2004); 3(11): 935-949.
- Lengauer T, Rarey M. Computational methods for biomolecular docking. Current opinion in structural biology, (1996); 6(3): 402-406.
- Śledź P, Caflisch A. Protein structure-based drug design: from docking to molecular dynamics. Current opinion in structural biology, (2018); 4893-102.
- Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. Journal of molecular biology, (1982); 161(2): 269-288.
- Wang L, Wu Y, Deng Y, Kim B, Pierce L, et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. Journal of the American Chemical Society, (2015); 137(7): 2695-2703.
- Zhang M-Q, Wilkinson B. Drug discovery beyond the ‘rule-of-five’. Current opinion in biotechnology, (2007); 18(6): 478-488.
- Giménez B, Santos M, Ferrarini M, Fernandes J. Evaluation of blockbuster drugs under the rule-of-five. Die Pharmazie-An International Journal of Pharmaceutical Sciences, (2010); 65(2): 148-152.
- Tripathi P, Ghosh S, Talapatra SN. Bioavailability prediction of phytochemicals present in Calotropis procera (Aiton) R. Br. by using Swiss-ADME tool. World Scientific News, (2019); 131147-163.
- Alshabrmi FM, Alkhayl FFA, Rehman AJEJoP. Novel Drug Discovery: Advancing Alzheimer's Therapy through Machine Learning and Network Pharmacology, (2024); 176661.
- Rehman A, Fatima I, Wang Y, Tong J, Noor F, et al. Unveiling the multi-target compounds of Rhazya stricta: discovery and inhibition of novel target genes for the treatment of clear cell renal cell carcinoma, (2023); 165107424.
- Lohohola PO, Mbala BM, Bambi S-MN, Mawete DT, Matondo A, et al. In silico ADME/T properties of quinine derivatives using SwissADME and pkCSM webservers, (2021); 42(11): 1-12.
- Rehman A, Noor F, Fatima I, Qasim M, Liao MJCiB, et al. Identification of molecular mechanisms underlying the therapeutic effects of Celosia Cristata on immunoglobulin nephropathy, (2022); 151106290.
- Mishra S, Dahima RJJodd, therapeutics. In vitro ADME studies of TUG-891, a GPR-120 inhibitor using SWISS ADME predictor, (2019); 9(2-s): 366-369.
- Aloufi B, Alshabrmi FM, Sreeharsha N, Rehman AJJoBS, Dynamics. Exploring therapeutic targets and drug candidates for obesity: a combined network pharmacology, bioinformatics approach, (2023); 1-22.