

Full Length Research Article
Bioinformatics-Driven Identification of Genetic Biomarkers and Therapeutic Targets in Dengue Virus Infection
Mohd Imran1,2, Mashael N. Alanazi1, Howayada Mahany Mostafa3, Abdullah R. Alzahrani4, Amer Ali Alamri5, Kholood Mohammed Moafa6, Feras Salah Albasha7, Latifa Fahad Almohsen8, Nader Sulaiman Ayyat Alanazi9, Mawahib Hassan Dirar Mokhtar10, Abuzer Ali11, Abida Khan1,2*
Adv. life sci., vol. 12, no. 1, pp. 211-223, February 2025
*- Corresponding Author: Dr Abida Khan (Email: aqua_abkhan@yahoo.com)
Authors' Affiliations
2.Center for Health Research, Northern Border University, Arar – Saudi Arabia
3. Department of Chemistry, College of Science and Arts-Rafha, Northern Border University, Arar – Saudi Arabia
4. Department of Pharmacology and Toxicology, Faculty of Medicine, Umm Al-Qura University, Al-Abidiyah, P.O.Box 13578, Makkah, 21955 – Saudi Arabia
5. The University Medical Center, Taibah University, Medina 41411 – Saudi Arabia
6. Department of Patients Services, Armed Forces Hospital, Jazan 45911 – Saudi Arabia
7. Department of Inpatient Pharmacy, Dr. Sulaiman Alhabib Hospital, Shafah Branch, Riyadh 12933 – Saudi Arabia
8. College of Clinical Pharmacy, King Faisal University, Al-Ahsa 36291 – Saudi Arabia
9. Department of Pharmacy, King Salman Specialist Hospital, Hail 55436 – Saudi Arabia
10. Harris Teeter Pharmacy, North Carolina 28412 – United States of America
11. Department of Pharmacognosy, College of Pharmacy, Taif University, P.O. Box 11099, Taif 21944 – Saudi Arabia
[Date Received: 24/08/2023; Date Revised: 30/07/2024; Date Published: 31/12/2024]
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: The Dengue virus (DENV) poses a considerable worldwide health threat, with its pathogenic processes not yet fully understood. Identifying biomarkers and elucidating the molecular underpinnings of dengue fever could enhance diagnostic and treatment strategies.
Methods: The study utilised bioinformatics methods to examine differentially expressed genes (DEGs) linked to dengue illness. Gene expression profiles from dengue-infected and normal samples were compared using datasets GSE51808 and GSE176079. Gene ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway enrichment studies, protein-protein interaction (PPI) network development, and miRNA/transcription factor analyses were conducted. Furthermore, protein-drug interaction studies and molecular docking were employed to corroborate therapeutic potential.
Results: A total of 555 differentially expressed genes (DEGs) were found in dengue-infected samples from GSE51808, including 174 upregulated genes, whereas 812 DEGs were identified in GSE176079, including 71 upregulated genes. Enrichment analysis identified significant pathways and activities related to these genes. Ten hub genes, namely SLC4A1, SNCA, TMOD1, and EPB42, were recognised as pivotal to dengue pathogenesis. Molecular docking confirmed interactions between approved drugs and hub genes: Atenolol and Metoprolol exhibited robust binding to SLC4A1, with binding scores of -6.478 and -6.032 kcal/mol, respectively, whereas Ketoconazole and Gentian violet interacted with SNCA, yielding binding scores of -6.2 and -6 kcal/mol.
Conclusion: The study emphasises the efficacy of bioinformatics in identifying biomarkers and treatment targets for dengue disease. SLC4A1 and SNCA were recognised as prospective indicators, whereas Atenolol, Metoprolol, Ketoconazole, and Gentian violet surfaced as promising treatment possibilities.
Keywords:Dengue fever virus (DENV); Gene Ontology (GO); Protein-Protein Interaction (PPI); Pathway Enrichment Analyses; Differentially Expressed Genes (Degs)
Introduction![]()
Dengue virus disease, also referred to as dengue fever, is an infectious disease caused by one of the dengue virus's four serotypes and transmitted by mosquitoes. This is a prominent worldwide health concern in regions with tropical and subtropical climates. Aedes mosquitoes, specifically Aedes aegypti, are the primary vectors for transmitting the disease. Dengue Hemorrhagic Fever (DHF) is an intensified form of dengue fever.
The spectrum of symptoms varies from mild dengue fever to fatal dengue shock syndrome. Currently, no scientifically established vaccine is accessible, which presents a notable public health issue, especially in tropical and subtropical areas [1,2]. Furthermore, the dengue complex comprises four DENV serogroups that are antigenically similar to one another but distinct from one another: DENV-1, DENV-2, DENV-3, and DENV-4, which are formed by dengue viruses (DENVs) of the genus Flavivirus, family Flaviviridae [3].
Dengue causes over 400 million illnesses and 22,000 fatalities annually. Predominantly, it has been recorded in over 100 nations. DENV, an enveloped RNA virus with a positive strand, is primarily transmitted by Aedes mosquitoes. NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 are the seven non-structural proteins that are found in this virus, which is a member of the Flaviviridae family of positive-strand RNA viruses [4,5]. A recent WHO study states that DENV is now a significant virus-caused illness following COVID-19 due to the rising number of infected individuals in 2020 [6].
Annually, the dengue virus infects about 390 million people worldwide; a 95% credible interval suggests that number could range from 284 million to 528 million. Out of them, 96 million individuals, with a 95% respectable range of 67 million to 136 million, show various levels of severity in clinical symptoms [6]. In the field of DHF study, molecular diagnostic methods played an important role in mapping inclusive gene expression profiles [7-10].
The approaches presented in this study have successfully found unique gene expression patterns that exhibit high accuracy in diagnosis, surpassing 95% in specific scenarios [11]. The precise identification of gene expression profiles showed tremendous potential for increasing the early diagnostic practices for DHF. Identifying these patterns in the early stages of infection can enhance the accuracy of the prognosis, which might give rise to better ways of managing and treating them. This method shows how important molecular diagnostics are for understanding and treating infectious diseases like DHF, where rapid and precise diagnosis is crucial in obtaining good patient results.
The application of computational biology and systems biology approaches has significantly updated the drug discovery process, significantly decreasing the expenditures associated with medication development. As it is known, RNA microarray analysis is a highly efficient, effective, and reliable method for quantifying the expression of all genes. RNA microarrays have been used to profile the expression of many genes in dengue; however, specific genes still need to be studied thoroughly [12,13].
Moreover, in the past few years, differential expression analysis has become an increasingly prevalent bioinformatics method in cancer research. Differentially expressed genes (DEGs) can assist with establishing important diagnoses and finding effective treatments for dengue, both of which are necessary to stop the spread of the illness and prevent it from spreading [14-18].
The present study applied an extensive computational bioinformatics methodology to identify possible genes associated with dengue infection. The study explored the microarray datasets obtained from the Gene Expression Omnibus (GEO) database by applying various computational biology and bioinformatics approaches. This study retrieved and evaluated two microarray gene datasets from the GEO. DEGs were examined among the healthy and dengue-infected cells. Moreover, to clarify the molecular processes that cause dengue's progression and development, gene ontology, enrichment, and protein-protein interaction network analysis were used.
The study examined the differential expression of genes, miRNAs, and transcription factors in dengue-infectious cell lines. Moreover, the study sought to repurpose therapeutic drugs that are efficacious against dengue virus (DENV). Further, molecular docking confirmed the binding affinity of the drug with the identified proteins that may interact with the dengue virus or affect the infection.
The objective of this study is to find potential biomarkers linked to dengue infection through bioinformatics methods. This project aims to explain the molecular underpinnings of DENV progression by analysing microarray gene expression datasets from the Gene Expression Omnibus (GEO) database, identifying important genes, miRNAs, and transcription factors, and repurposing therapeutic medicines for DENV treatment. The study utilises molecular docking to confirm drug-protein interactions, providing insights into possible therapeutic approaches for dengue disease.
Methods![]()
Dataset Retrieval
The GEO database (https://www.ncbi.nlm.nih.gov/geo/) provided two gene expression profiles, GSE51808 and GSE176079, which were collected using microarray methods [19]. The scientific community submits functional genomic data sets obtained from next-generation sequencing and high-throughput microarray technology to GEO, an international public repository.
The datasets GSE51808 and GSE176079 were selected for their significance to elucidating the molecular mechanisms of dengue virus (DENV) infection and being consistent with the study's aims. GSE51808 offers a thorough examination of gene expression alterations in whole-blood samples from individuals with dengue fever (DF) and dengue hemorrhagic fever (DHF) during both acute and recovery stages, providing significant insights into the systemic immune response to DENV. Moreover, it encompasses samples from diverse clinical severities, enabling an extensive investigation of gene expression patterns linked with the disease. Conversely, GSE176079 concentrates on monocyte subsets isolated from peripheral blood, facilitating an in-depth examination of cell-type-specific immune responses These two datasets were generated from GPL13158 (Affymetrix HT HG-U133+ PM Array Plate) and GPL21290 (Illumina HiSeq 3000) platforms.
The dataset labelled GSE51808 employs a systems biology methodology to examine the innate immune response to dengue virus (DENV) infection in whole-blood samples from 28 individuals diagnosed with dengue (DF n = 18, DHF = 10). In addition to the specimens obtained during the initial nine days of symptoms (acute illness), samples from 19 patients (six with DHF and thirteen with DF) were collected during recuperation four weeks after discharge. Blood samples from 9 non-infected, healthy donors were also utilized as control subjects for immunological and transcriptomic analyses.
The other dataset with accession number GSE176079 involves sorting monocyte subsets from peripheral blood cells. The sorting process excluded CD3, CD19, CD20, CD56, CD66b, and NKp30 positive cells. This work analyzes various peripheral blood monocyte subsets to provide a thorough grasp of their properties and roles. The cells were classified into DENV and healthy cells for this analysis. In order to ensure the reliability of the data, both datasets were subjected to a quality evaluation prior to analysis.
Platform compatibility, sample integrity, normalisation, and reproducibility comprised the data preparation procedures. These procedures ensured that the datasets were reliable and appropriate for subsequent bioinformatics analyses, thereby improving the validity and reliability of the study's findings.
Differentially Expressed Genes (DEGs) Analyses
The differentially expressed genes (DEGs) analysis was performed on the GEO2R platform. The DEGs between healthy and infected Dengue Virus (DENV) were examined and shown via the GEO2R platform.
GEO2R, a web-based tool offered by the Gene Expression Omnibus (GEO), was selected for differential gene expression analysis due to its use of the limma package, a well-established statistical approach for detecting differentially expressed genes (DEGs) in microarray datasets. GEO2R provides an intuitive interface and comprehensive preprocessing functionalities, facilitating normalisation and statistical modifications (e.g., corrections for multiple testing) to guarantee dependable outcomes.
The technology is especially adept at comparing numerous conditions within a dataset, as necessitated by this study. Limma programs from the Bioconductor project were applied to identify as significant DEGs in the GSE51808 and GSE176079 datasets those genes exhibiting a |log2fold change (FC)| ≥ 2 and an adjusted P < 0.05 [20]. The threshold of |log2FC| > 2 was selected to emphasise genes exhibiting significant expression changes, indicative of physiologically relevant modifications rather than minor fluctuations. This threshold facilitates the discovery of genes that play crucial roles in the pathogenesis of the disease, perhaps implicated in essential processes such as immune response, viral replication, or inflammation.
The adjusted p-value < 0.05 was utilised to mitigate false positives in high-throughput data analysis, hence augmenting the dependability of the outcomes. DEGs were visually represented through volcano plots, which were produced using the ggplot2 package in the R programming environment (version 3.6.3). Concurrently, Microsoft Excel was used to generate Venn diagrams depicting the overlap of DEGs from the GSE51808 and GSE176079 datasets.
Protein-Protein Interaction Formulation
The STRING database (Version 12.0) has been used as a search tool to retrieve interacting genes and generate a protein-protein interaction (PPI) network, demonstrating the connections between the specified genes. Protein-protein interactions, encompassing both functional linkages and physical interactions, are methodically gathered and integrated by the STRING database [21]. The most significant possible score for a confidential interaction—0.9—was attained. Using Cytoscape software (An open-source graph library built using JavaScript called Cytoscape.js).
Cytoscape was employed for the visualisation and analysis of protein-protein interaction (PPI) networks of differentially expressed genes (DEGs). Cytoscape was selected for its versatility and capacity to integrate many data sources, including STRING for protein-protein interaction data and supplementary functional annotations. The broad selection of plugins, such as MCODE for network cluster identification and ClueGO for route enrichment visualisation, facilitates thorough research of gene-gene relationships and biological pathways.
The most typical application for it is as a component of visualization software, which enables the rendering of interactive graphs in web browsers; the PPI network was further visualized, and the hub protein was discovered [22].
Gene Ontology (GO) and Pathway Enrichment Analysis
The g:Profiler tool was used to conduct GO and KEGG pathway enrichment analysis for the common DEGs [23]. The g:Profiler program is a comprehensive and frequently updated resource for functional enrichment analysis. Gene Ontology (GO), the Kyoto Encyclopedia of Genes and Genomes (KEGG), and TRANSFAC are all part of this all-inclusive toolset that allows for detailed analysis of gene assemblies.
This tool was selected for its capacity to provide extensive functional annotations and pathway insights by synthesising data from various meticulously curated biological sources. The intuitive interface and comprehensive statistical framework for enrichment analysis guarantee accurate identification of biological processes, cellular components, and molecular functions linked to the DEGs. Molecular Functions (MF), Cellular Components (CC), and Biological Processes (BP) were the specific dimensions used in this study.
Furthermore, the KEGG pathway enrichment analysis was carried out using the Gene Enrichr web platform, a crucial resource for understanding the functional consequences of shared DEGs in particular biological pathways.
Relationships between DEG microRNAs and transcription factors
The bioinformatics prediction tool miRNA Data Integration Portal was used to determine the type of relationship between the miRNA and the selected DEGs [24]. This bioinformatics tool was selected for its ability to integrate data from multiple sources to predict miRNA-target interactions, facilitating a comprehensive knowledge of post-transcriptional regulatory mechanisms.
Its capacity to delineate miRNA interactions with DEGs offered essential insights into the regulatory networks that may be altered during DENV infection. Better knowledge of miRNA-gene interactions is attainable because of the program's capacity to integrate data from 26 distinct miRNA databases. Another objective of this study was to determine the relationship between transcription factors and DEGs by analyzing TRRUST, which is a database of human and mouse transcriptional regulatory networks that have been manually curated [25].
The TFs-DEGs interaction network and the miRNAs-DEGs interaction network were built by the miRNA-centric network visual analytics platform or miRNet tool [26].
Identification of small molecule drug
The DGIdb (https://www.dgidb.org/), an online resource that compiles information on drug-gene interactions from over 30 separate databases, was used to screen small molecules. The Drug-Gene Interaction Database (DGIdb) was utilised to identify small compounds that interact with the specified differentially expressed genes (DEGs).
DGIdb aggregates data from more than 30 unique databases, rendering it a valuable resource for investigating established drug-gene interactions and druggable targets. The comprehensive database and strong filtering capabilities enable researchers to identify FDA-approved medications or investigational substances suitable for repurposing. The objective was to identify therapeutic compounds that exhibited specificity towards the DEGs that had been pre-selected.
The metadata on drug-gene interactions and gene druggability that is compiled, organized, and displayed in this database comes from various sources, including publications, databases, and online sources [27]. The potential therapeutic drugs that target the (DEGs) have been identified using the interaction score.
Molecular docking
Drug development relies heavily on the use of molecular docking as a key technique. By using this method, it is possible to model the interactions between small molecules and proteins on an atomic scale, which helps to shed light on significant biochemical processes and classify the behaviour of small molecules within the binding regions of target proteins [28].
The 3D structure of the proteins was retrieved from the protein data bank database [29]. The present study used Autodock (version -1.5.7) to prepare the protein structures. The process involved removing the water molecules from the protein, integrating the missing atoms, and introducing hydrogen atoms and Kollman charges to every atom. Further, the derived protein structure was saved in the PDBQT format. A grid box was constructed in proximity to the binding pocket. Moreover, specific docking parameters were standardized to optimize the results.
The number of modes was fixed to 20, the exhaustiveness was set to 100 and an energy difference of 4kcal/mol was employed to maximize the favourable binding interactions. The identified proteins underwent molecular docking with AutoDock Vina with their respective interacting drugs [30]. The process successfully identified potential candidates with high binding affinity.
Results![]()
Comparing Infected and Healthy Cells for DEGs
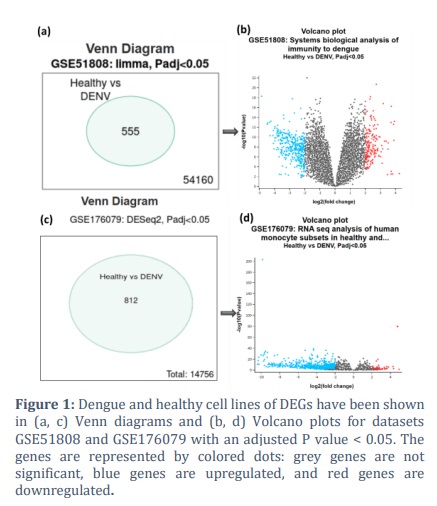
Through analysis of two microarray-based gene expression profiles, GSE51808 and GSE176079, differentially expressed genes between DENV and healthy cells were identified. GSE51808 involved 9 healthy and 47 infected cells (DENV), whereas GSE176079 had 9 healthy and 17 infected cells. GEO2R analysis identified a total of 555 genes in GSE51808 and 812 genes in GSE176079 as significant with an adjusted P < 0.05, as depicted in Figure 1 (a, c). Genes with |Log2FC|≥2 were regarded as DEGs among these significant genes.
A volcano plot was created to show the DEGs between healthy and DENV, shown in Figure 1(b, d). The top 100 DEGs sorted by corrected P values from each dataset were selected for additional investigation. To identify the shared DEGs between two datasets, GSE51808 and GSE176079, the Venn diagram depicted in supplementary figure S1 was constructed. Five were upregulated, and ninety-five were downregulated among the frequent DEGs associated with GSE51808 and GSE176079. These common five genes, namely OLIG1, RTN1, PID1, FCER1A, and ANKH, exhibited upregulation. The visual representation known as the volcano plot summarises a differential expression analysis conducted on the DENV-infected and healthy individuals, according to the datasets GSE51808 and GSE176079.
Supplementary Files S2 and S3 listed the genes that were downregulated and upregulated for GSE51808 and GSE176079, respectively. Figure 1 (b, d) presents volcano plots that depict the differential gene expression between DENV-infected and healthy samples, emphasising important genes with |Log2FC| > 2 and an adjusted p-value < 0.05. In GSE51808, a balanced distribution of upregulated and downregulated genes signifies a dynamic immune response, where elevated genes presumably denote antiviral pathways and downregulated genes suggest host regulatory systems.
In contrast, GSE176079 exhibits a majority of downregulated genes, indicating substantial transcriptional repression in monocyte subsets, which may signify viral immune evasion tactics or cell-type-specific host responses. The Venn diagrams (Figure 1a, c) illustrate common differentially expressed genes (DEGs), comprising five consistently elevated genes—OLIG1, RTN1, PID1, FCER1A, and ANKH—that may represent strong possibilities for biomarker or therapeutic advancement.
The results, coupled with the protein-protein interaction (PPI) network analysis, highlight the significance of these DEGs in host-pathogen interactions and their prospective functions in immune regulation and viral propagation. The analysis utilized an adjusted P-value threshold of less than 0.05. The plot indicates several significantly differentially expressed genes in both upregulated (red) and downregulated (blue) categories, which could be candidates for further investigation into their roles in the immune response to dengue infection.
The genes in the extremes of the plot (far right and far left) with high significance (top of the plot) are of particular interest as potential key players in the pathophysiological response to DENV. Grey dots represent genes that are not significantly differentially expressed or do not meet the threshold criteria. A more even distribution of upregulated and downregulated genes was observed in the first dataset (GSE51808), in contrast to the heavily downregulated genes observed in the second dataset (GSE176079).
The even distribution of DEGs in GSE51808 suggests a strong and intricate reaction to DENV infection, which may encompass various cellular pathways and processes. However, the dataset GSE176079's downregulation dominance points to gene expression suppression, which may reflect a distinct component of the immune response or a response unique to certain cell types to DENV.
In total, the data from both sets provide insight into the host response to DENV infection. The GSE51808 dataset shows a mixed regulatory response to DENV, with notable upregulation and downregulation, indicating that cells actively engaged in repression and activation mechanisms. On the other hand, the GSE176079 dataset displays a strong pattern of downregulation, which may indicate a more focused or muted reaction. These genes that are upregulated may generate proteins that play a crucial role in slowing the virus's fast growth and regulating the host's immune response.
Additionally, these genes can shed light on the molecular mechanisms the virus uses for replication, which is essential for developing strategies to block these viral-host interactions. On top of that, by using existing drugs, the proteins produced by elevated genes could be used as potential targets for repurposing medications, speeding up the development of effective treatments for DENV.
Thus, the study aimed to analyze the GSE51808 and GSE176079 datasets for their upregulated DEGs.
Protein-Protein Interaction Network
Comparing the GSE51808 and GSE176079 datasets, which contain networks of protein-protein interactions (PPIs) built from upregulated differentially expressed genes (DEGs), revealed clear structural differences. In supplementary figure S2, the upregulated PPI networks of the DEGs were displayed. Numerous significant hubs in the complex and interconnected PPI network were linked to the 174 upregulated DEGs from GSE51888. After ALAS2, DMTN, and SNCA, most of the nodes were found in EPB42 and SLC4A1. Moreover, a rational number of nodes were observed in STRADB, DCAF12, HBQ1, TMOD1, SLC25A39, OSBP2, ANK1, and TRIM58. The PPI networks of the 71 upregulated DEGs in the GSE176079 dataset differed greatly. It failed to create a robust PPI network with notable nodes and exhibited no signs of complicated network dynamics. Due to its inherent complexity and abundance of hubs, the GSE51808-PPI network is an appropriate candidate for additional investigation because of the profound biological insights it may provide. On the other hand, the GSE176079 network was not used further because its structure was not very advanced to construct a PPI network.
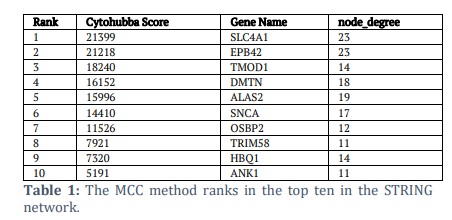
The top 10 hub genes were found after further investigation of the GSE51808 PPI network with the help of CytoHubba [31]. It is a Cytoscape plugin developed to locate important nodes in biological networks. The genes identified include SLC4A1, EPB42, TMOD1, DMTN, ALAS2, SNCA, OSBP2, TRIM58, HBQ1, and ANK1 as shown in supplementary figure S3. A color gradient ranging from red to yellow was used to visualize the hubs in Cytoscape, indicating their relative significance in the network. Its Maximal Clique Centrality (MCC) and Degree values were used to determine the gradient. As the MCC/Degree values decrease, the color changes from red to yellow. By applying the MCC method to the nodes in the biggest cliques of the network, the study was able to identify highly connected genes that are likely essential for many biological processes and functions. At the same time, the Degree method offered a supplementary viewpoint by measuring the number of direct connections each gene has, drawing attention to genes that are important hubs in the network because of the number of times they interact with each other. It identifies key genes in the network, which may lead to new avenues of research or treatments. In addition, the STRING network's top ten genes, as ranked by the MCC method, are displayed in Table 1. Here, SLC4A1 and EPB42 showed the highest number of nodes with a degree of 23, and the highest cytohubba score was for SLC4A1, as also shown by the color gradient. Further, TMOD1 and DMTN showed the next highest cytohubba scores, followed by ALAS2, SNCA, and OSBP2.
These genes are highly interconnected, highlighting their importance in cellular processes disrupted by DENV. SLC4A1 and EPB42 play roles in maintaining ionic balance and erythrocyte stability, potentially mitigating metabolic acidosis and haematological abnormalities like thrombocytopenia seen in dengue. TMOD1 and DMTN are involved in cytoskeletal organization, which DENV exploits for replication. ALAS2, key in heme biosynthesis, may support impaired red blood cell function during infection. SNCA and OSBP2 are linked to oxidative stress and lipid metabolism, suggesting roles in DENV replication and systemic damage. TRIM58 and HBQ1 contribute to protein regulation and red blood cell function, while ANK1 supports erythrocyte cytoskeletal stability and may respond to hypoxia during severe dengue. These hub genes provide insights into DENV’s impact on host cellular pathways and highlight potential therapeutic targets.
Comparative analysis of DEGs' gene ontology and pathway enrichment
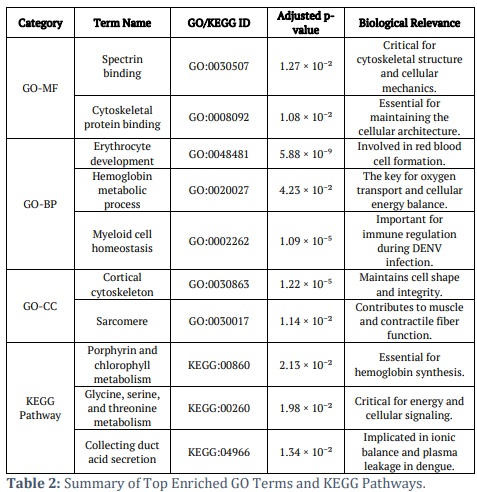
Gene Ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway enrichment analyses were carried out with the assistance of the g:Profiler tool. These studies contributed to explaining the biological significance of the upregulated DEGs found in the GSE51808 datasets. This approach aimed to better understand the possible roles of these DEGs in the studied biological systems by elucidating their functional and pathway associations.
The GO analysis targeted three areas: cellular components (CC), biological processes (BP), and molecular functions (MF), as shown in supplementary figure S4 (a). The upregulated DEGs were highly enriched in spectrin and cytoskeletal protein binding functions within the MF domain. According to these results, these genes play a crucial role in cellular architecture's structural and mechanical components. Findings from the study implicate these DEGs in a wide variety of BP. These include controlling protein polymerization, maintaining and specializing myeloid cells, and forming and specializing red blood cells. Furthermore, these genes seem to be involved in important physiological processes, such as developing red blood cells (RBCs) and maintaining homeostasis in multicellular organisms. They are also involved in more specific pathways, such as metabolizing haemoglobin and regulating the assembly of protein-containing complexes negatively. The diverse processes point to the common DEGs playing a significant role in haematopoiesis and related cellular mechanisms. The frequent DEGs identified in the CC analysis are related to several structural and organizational components of the cell. These components include the cortical cytoskeleton, cell cortex, spectrin-associated cytoskeleton, and supramolecular structures such as fibres and polymers. Their connection to sarcomeres, myofibrils, and contractile fibres suggests a possible involvement in muscle function and structure.
In addition to the GO analysis, the KEGG pathway enrichment study provided insight into the participation of these DEGs in particular metabolic and regulatory processes, as shown in supplementary figure S4 (b). The most enriched pathways comprised porphyrin and chlorophyll metabolism, essential for haemoglobin formation, and glycine, serine, and threonine metabolism, vital for energy production and cellular signalling. These findings underscore the influence of DENV infection on metabolic systems essential for cellular and systemic homeostasis. Furthermore, pathways including collecting duct acid secretion were enriched, indicating potential disturbances in ionic equilibrium and pH regulation, which may lead to clinical manifestations such as plasma leakage and organ failure in severe dengue. Collectively, these findings underscore the role of elevated DEGs in processes and pathways essential to haematopoiesis, metabolism, and cellular structural integrity. This highlights their possible involvement in the pathogenesis of DENV infection, establishing a basis for focused study on therapeutic approaches.
Hence, these thorough studies offer an in-depth comprehension of the specific and combined functions of the shared DEGs from GSE51808 and emphasize the complex network of biological functions and pathways in which they participate. This provides a basis for further investigation into their functions in the physiological setting and potential consequences in disease processes or treatment strategies. The KEGG pathway enrichment analysis elucidated the biological significance of these DEGs, as illustrated in supplementary figure S4 (b). Table 2 summarises the principal Gene Ontology (GO) keywords and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways to explain the enrichment analysis.
Micro RNA, transcription factor, and DEG interaction prediction
Supplementary Figure S5 shows the result of using miRNet tools to build a network of interactions between miRNAs and DEGs and between TFs and DEGs. Blue stands for gene-microRNA (miRNA), green for transcription factors, and yellow for differentially expressed genes (DEGs) (TFs). The mirDIP database identified multiple miRNAs that interact with the DEGs. Utilizing the TRRUST database, several transcription factors (TFs) were identified, suggesting that they may control the expression of DEGs. Through the use of miRNet, the interconnections between genes and miRNAs, as well as TFs and DEGs, were visualized and analyzed. Supplementary Figure S5 shows the complex relationship between the regulators of biological systems regarding gene expression by various diseases.
Hub genes found to be significant in the analysis are SLC4A1, EPB42, and ANK1. Their potential interactions with TFs (HIF1A) and multiple miRNA connections indicate that these hubs play an important role in the regulatory pathways. Regarding the number of interactions between DEGs and miRNAs, SLC4A1 had the most, followed by ANK1 and EPB42. Interactions with microRNAs were observed with other DEGs, including OSBP2, ALAS2, SNCA, TMOD1, DMTN, and TRIM58. The miRNA, hsa-mir-34a-5p, showed interaction with EPB42, ANK1, ALAS2, SNCA, TMOD1, DMTN, and TRIM58 genes. Here, ALAS2 interacted with a microRNA and two transcription factors (TFs), one of which was HIF1A. In addition, evidence suggests that the transcription factor HIF1A controls ANK1 gene expression. These microRNAs and transcription factors could be essential for developing and progressing dengue infections. These networks are very helpful for understanding diseases at the molecular level, finding key regulatory elements that could be used as therapeutic targets, and finding biomarkers that can be used for diagnosis or prognosis. One of the most important layers for regulating gene expression after transcription is the presence of miRNA nodes. Network analysis is important when studying biological conditions to gain insight into the regulatory cascades.
The hub genes noticed in this study—SLC4A1, EPB42, and ANK1—are integral to pathways altered during dengue virus (DENV) infection. SLC4A1 is an essential element of anion exchange and ionic homeostasis, with its dysregulation likely associated with metabolic acidosis and ionic imbalances noted in severe dengue infections. Its strong interactions with miRNAs indicate it is a crucial post-transcriptional regulatory hub during infection. EPB42 is crucial for preserving erythrocyte membrane integrity, and its regulation by hsa-mir-34a-5p suggests a function in mitigating oxidative and mechanical stress caused by DENV. ANK1, regulated by the transcription factor HIF1A, plays a role in cytoskeletal organisation and erythrocyte stability. The disruption of these systems aligns with thrombocytopenia, haemolysis, and vascular leakage noted in dengue pathogenesis. The interactions of these hub genes with miRNAs, especially hsa-mir-34a-5p, indicate possible treatment opportunities. Modulating miRNA activity may restore gene expression equilibrium, hence alleviating illness severity. The transcription factor HIF1A, which regulates both ANK1 and ALAS2, underscores a connection among hypoxia, inflammation, and erythropoiesis, offering further avenues for therapeutic intervention. The findings highlight the translational potential of the identified hub genes and their regulatory networks, facilitating the development of new diagnostic biomarkers and targeted therapies for dengue fever.
Protein-Drugs analysis
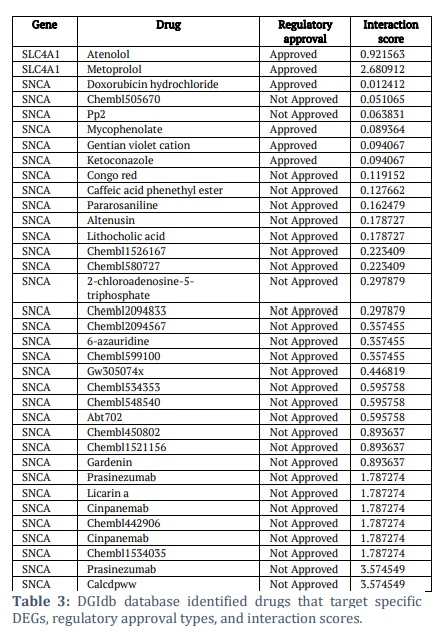
Repurposing drugs for novel diseases has advanced significantly through the identification of crucial differentially expressed genes (DEGs) as prospective therapeutic targets. In this case, a strategic approach to identifying drugs that may interact with these gene products involves selecting the 10 DEGs from a database like DGIdb. A thorough search was carried out in the DGIdb database on various drugs regarding 10 important DEGs that were selected as possible therapeutic targets. Many important factors involving ANK1, SLC4A1, EPB42, TMOD1, DMTN, ALAS2, SNCA, OSBP2, TRIM58, and HBQ1 were identified in the study. This provides enough evidence to suggest that these genes may serve as targets in the future. DGIdb analysis predicted the interaction between the drugs and specific proteins. Additional investigation into the DGIdb database unveiled 33 candidate drugs for SNCA and 2 candidate drugs for SLC4A1. Drugs such as Metoprolol and Atenolol were found to target SLC4A1. In contrast, Doxorubicin hydrochloride, Gentian violet, Ketoconazole, Mycophenolic acid, Congo red, Caffeic acid phenethyl ester, Pararosaniline, Altenusin, and Lithocholic acid targeted SNCA based on the interaction score (Table 3). For SLC4A1, Atenolol and Metoprolol were found to be approved drugs with an interaction score of 0.921563 and 2.680912, respectively. For SNCA, four approved drugs were found, namely Doxorubicin hydrochloride, Mycophenolate, Gentian violet cation, and Ketoconazole, with interactive scores of 0.012412, 0.089364, 0.094067, and 0.094067. A molecular docking analysis was conducted to validate the effectiveness of these small-molecule pharmaceuticals further.
In the repurposing effort, the prediction of protein-drug interactions and the subsequent identification of candidate drugs for SLC4A1 and SNCA are critical. The fact that drugs like Metoprolol and Atenolol, which are typically used for cardiovascular indications, have been found to target SLC4A1, suggesting an unexplored therapeutic potential in the context of DENV infection [32]. Because of this, the intriguing prospect that these medications may affect cellular environments in ways that are useful for treating viral infections is now being considered. Interaction scores help quantitatively rank potential drugs based on their target protein effects. Since these medications have previously been tested for safety and effectiveness, their approval status is a key consideration that could speed up moving them into clinical trials for new indications. Approved drugs such as doxorubicin hydrochloride and ketoconazole were among those identified, lending credibility to the idea that current medications could be repurposed for new therapeutic uses, according to SNCA. Atenolol and metoprolol have encouraging interaction scores with SLC4A1, indicating a high likelihood of repurposing.
In addition, molecular docking analysis simulates the molecular interactions between the target proteins and the small-molecule medications, providing additional validation. The comprehensive approach to identifying new uses for existing drugs and speeding up the process of finding effective disease treatments involves exploring DEGs as drug targets using databases like DGIdb, followed by molecular docking and further validation.
Molecular Docking
Studies of molecular docking were carried out on the selected genes SLC4A1 and SNCA, which are represented by the PDB IDs 4YZF and 1XQ8, respectively [33,34]. These studies were conducted to identify potential small-molecule inhibitors that could be used in therapeutic interventions. After conducting an exhaustive analysis of the binding pockets of every protein, it was possible to generate accurate docking grids. The dimensions of the grids were carefully chosen to encompass the active sites effectively. For SLC4A1, a grid measuring 18 × 18 × 18 Å3 was established, centered at coordinates (9.331, 2.298, 41.941) Å. Similarly, for SNCA, a grid measuring 20 × 20 × 20 Å3 was determined, centered at coordinates (91.237, -21.517, -28.3) Å. The spatial parameters play a crucial role in ensuring that the docking simulations appropriately represent the physical limitations of the molecular interactions occurring within the functional domains of the proteins.
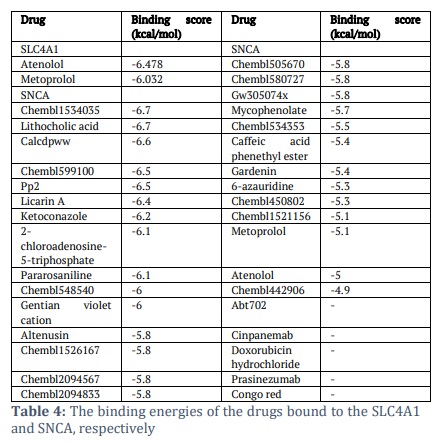
All identified drug candidates (33 for SNCA and 2 for SLC4A1) were subjected to docking against SNCA and SLC4A1, respectively. Table 4 shows the binding score for drug candidates. The docking procedure resulted in identifying Atenolol and Metoprolol as the primary interactors for SLC4A1. They exhibited binding scores of -6.478 and -6.032 kcal/mol, respectively, indicating a strong affinity and possible inhibitory ability. Simultaneously, SNCA interactors, including Chembl1534035 and Lithocholic acid, exhibited binding energies of -6.7 kcal/mol, while Calcdpww, Chembl599100, and Pp2 had binding scores ranging from -6.5 to -6.6 kcal/mol. Licarin A, Ketoconazole, 2-chloroadenosine-5-triphosphate, Pararosaniline, Chembl548540, and Gentian violet cation also showed binding scores ≤ -6 kcal/mol. The binding scores reported suggest strong molecular interactions that may result in considerable biological impacts. Binding energies below -6 kcal/mol are typically regarded as strong, akin to established inhibitors that target analogous pathways. The interactions of Ketoconazole and Gentian violet with SNCA correspond with their established antifungal and antibacterial properties, indicating potential repurposing prospects for DENV-related disorders. The robust affinity of Atenolol and Metoprolol for SLC4A1 is significant, since their proven safety profiles as beta-blockers may facilitate therapeutic use in addressing DENV-induced metabolic disturbances. These findings highlight the clinical significance of the docking results and establish a basis for additional experimental validation for dengue virus targets. Among these, Ketoconazole and Gentian violet cation are the only approved drugs. The observed binding energies suggest strong interactions, which could have notable biological consequences.
Previous studies have investigated possible inhibitors of dengue virus (DENV) utilising computational and docking methodologies, establishing a framework for assessing the docking scores noted in this study. A study examined domain III of DENV across serotypes, investigating its interaction with the monoclonal antibody (mAb) 4E11. The docking (XP) values for the selected small molecules varied from -1.5 kcal/mol to -8.9 kcal/mol, signifying moderate to significant binding affinity (https://doi.org/10.1371/journal.pone.0311548). A separate study employed a computational method to evaluate a library of drugs approved by the FDA against the capsid protein of DENV, producing docking scores between -12.67 and -8.4 kcal/mol, indicating strong binding interactions (https://doi.org/10.1007/s11030-024-10980-z). In a distinct study, eight inhibitors of HCV RdRp were computationally evaluated against the co-crystallized structure of DENV-3 RdRp. After energy minimisation and XP docking, scores varied from -5.9 to 0.7 kcal/mol, indicating differing degrees of interaction strength (https://doi.org/10.1007/s11030-023-10716-5).
These investigations reveal a broad spectrum of docking scores, with potent inhibitors generally exhibiting scores more negative than -8 kcal/mol, whilst moderate inhibitors fall within the range of -6 to -8 kcal/mol. The binding affinities of the interactors found in this study Atenolol (-6.478 kcal/mol) and Metoprolol (-6.032 kcal/mol) for SLC4A1, and Ketoconazole (-6.2 kcal/mol) for SNCA are categorised as moderate. This emphasises their therapeutic promise while indicating the necessity for additional optimisation to improve their efficiency as DENV inhibitors. Experimental validation and structure-based optimisation of these compounds are essential further stages to ascertain their therapeutic efficacy against DENV infection.
The identification of Atenolol, Metoprolol, Ketoconazole, and Gentian violet as prospective pharmaceuticals via the DGIdb database underscores the possibility of repurposing existing medications for dengue therapy. These medications, possessing validated safety profiles and pharmacokinetics, provide an expedited route to clinical implementation. Atenolol and Metoprolol, widely utilised beta-blockers, exhibited interactions with SLC4A1, indicating a potential mechanism that may regulate ionic imbalances and alleviate metabolic disruptions induced by DENV infection. Likewise, Ketoconazole and Gentian violet, possessing antifungal and antibacterial characteristics, exhibited binding to SNCA, suggesting potential disruption of viral replication mechanisms. Nevertheless, although these results are encouraging, experimental validation is crucial to verify their antiviral effectiveness against DENV. Preclinical investigations, encompassing in vitro and in vivo experiments, are essential for evaluating efficacy, specificity, and possible mechanisms of action. Moreover, dosage optimisation is essential to reconcile therapeutic efficacy with safety, especially when elevated dosages are necessary for antiviral effectiveness. Understanding the stage at which these pharmaceuticals impede viral progression whether during entry, replication, or assembly will determine their therapeutic efficacy. Additionally, the exploration of combination therapies with current medications should be undertaken to improve efficacy and mitigate resistance risks. These pharmaceuticals may advance to clinical trials if preclinical outcomes are favourable, providing a solution to the urgent need for effective dengue treatments. This method highlights the effectiveness of drug repurposing in utilising existing resources to address new infectious illnesses.
Figures & Tables
This study used a comprehensive computational method that combined differential gene expression (DEG) analysis with the development of protein-protein interaction (PPI) networks to discover possible biomarkers for Dengue infection. The molecular mechanisms underlying the pathogenesis of Dengue were examined through the utilization of Gene Set Enrichment Analysis (GSEA), Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway analysis, and Gene Ontology (GO) enrichment analysis. In addition, by utilizing network pharmacology and in silico docking, we predicted potential therapeutic candidates that have a likely affinity for the identified DEGs. This study determined the clinical importance of SLC4A1 and SNCA concerning the severity of Dengue infection. These genes have been identified as prospective biomarkers that might differentiate between those infected with Dengue and those who are healthy. Because of this, they provide a novel method of carrying out diagnostic procedures. The microarray datasets GSE51808 and GSE176079 from the GEO database were analyzed, and the results showed that there was a detectable rise in the number of DEGs that were upregulated during Dengue infection. In this context, these genes likely play a part in regulating immune responses within the host and preventing the virus from spreading further.
Dataset GSE51808 stands out compared to GSE176079 due to the large number of upregulated DEGs. These genes can form a complex PPI network with many interaction hubs. The PPI analysis identified the top 10 hub genes, specifically SLC4A1, EPB42, TMOD1, DMTN, ALAS2, SNCA, OSBP2, TRIM58, HBQ1, and ANK1. It was found that SLC4A1, EPB42, and ANK1 played an essential role in the network of genes involving DEGs, miRNAs, and TFs, specifically with the miRNA hsa-mir-34a-5p and the transcription factor HIF1A. A prior study was compared, which examined dengue-infected samples using microarray data from datasets GSE28405, GSE38246, and GSE51808 that found 69 differentially expressed genes [17]. This study successfully identified the hub protein and made additional accurate predictions regarding the biomarkers IFI44L and IFI6 for DENV infection. These findings offer some possible targets for the potential treatment of dengue fever.
Further, in the current study, the DGIdb database provided additional predictions of protein-drug interactions, revealing 33 potential drugs that target SNCA and 2 potential drugs that target SLC4A1. These findings serve as a foundation for drug repurposing efforts. Molecular docking was used to enhance the search process, identifying 10 potential medication candidates for SNCA and 2 for SLC4A1. In terms of binding affinities, these candidates were the most promising. Among these, only two drugs were FDA-approved: ketoconazole and gentians violet cation for SNCA and Atenolol and metoprolol for SLC4A1,
Proteins encoded by the human SLC4A1 and SNCA genes are critical for the control of cellular physiology. Because of this protein, the SLC4A1 gene encodes anion exchangers, bicarbonate and chloride ions can pass through the cell membrane [35]. It is best known for its important role in red blood cells, where it controls the ratio of oxygen to carbon dioxide [36,37]. Certain disorders, including hereditary spherocytosis and hereditary elliptocytosis, have been associated with mutations in this gene. These diseases affect red blood cells in both structure and function [38]. The alpha-synuclein protein encoded by the SNCA gene is involved in synaptic function, neuronal plasticity, and adaptation [39]. The brain is the primary site of protein synthesis for this protein. Several diseases, such as multiple system atrophy, PD, and Lewy body dementia, affect the nervous system and are marked by alpha-synuclein accumulation [40,41]. These genes may experience an increase in expression levels in response to Dengue infection. Some of the host’s genes involve biological processes that the virus may affect or interact with. Research has indicated that alterations in the expression of certain host genes during Dengue infection may provide insight into the severity of the disease or identify possible areas for treatment intervention [42,43]. The virus may use its upregulation to improve its replication or transmission. Consequently, the host’s cells may halt the spread of dengue infection if the approved medications inhibit the genes.
Overall, the results show that computational bioinformatics is a powerful tool for unravelling the mysteries of dengue fever and its associated genetic and molecular alterations. In this study, new insights into Dengue's development have been found, and potential therapeutic targets include several important genes, such as SLC4A1, EPB42, ANK1, ALAS2, SNCA, TMOD1, DMTN, and TRIM58. The study also delves into understanding the complex host response to the Dengue virus, enhanced by the role of specific microRNAs and transcription factors, like hsa-mir-34a-5p and HIF1A, in gene regulation. The possibility of reusing currently licensed pharmaceuticals like Atenolol, Metoprolol, Ketoconazole, and Gentian violet to fight Dengue has been brought to light by the identifying of small-molecule drugs that strongly bind to important proteins like SLC4A1 and SNCA. Although these findings show potential, additional experimental validation is required to verify the effectiveness of these medications in fighting Dengue infection, thereby potentially facilitating the development of novel therapeutic approaches.
Supplementary Data
Readers may contact the corresponding author for supplemental data/information.
Acknowledgement
The authors extend their appreciation to the Northern Border University, Arar, KSA for funding this research work.
The authors declare that there is no conflict of interest regarding the publication of this paper.
![]() References
References
- Kok BH, Lim HT, Lim CP, Lai NS, Leow CY, et al. Dengue virus infection – a review of pathogenesis, vaccines, diagnosis and therapy. Virus Research, (2023); 324: 199018.
- Nanaware N, Banerjee A, Mullick Bagchi S, Bagchi P, Mukherjee A. Dengue Virus Infection: A Tale of Viral Exploitations and Host Responses. Viruses, (2021); 13(10): 1967.
- Murugesan A, Manoharan M. Dengue Virus. Emerging and Reemerging Viral Pathogens, (2020); 1: 281-359.
- Roy SK, Bhattacharjee S. Dengue virus: epidemiology, biology, and disease aetiology. Canadian Journal of Microbiology, (2021); 67(10): 687-702.
- Umareddy I, Chao A, Sampath A, Gu F, Vasudevan SG. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. The Journal of General Virology, (2006); 87(Pt 9): 2605-2614.
- World Health Organization. Dengue and severe dengue, (2024) Available at https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue (Accessed on July 1, 2024)
- Cordeiro MT, Silva AM, Brito CA, Nascimento EJ, Magalhães MC, et al. Characterization of a dengue patient cohort in Recife, Brazil. The American Journal of Tropical Medicine and Hygiene, (2007); 77(6): 1128-1134.
- Ubol S, Masrinoul P, Chaijaruwanich J, Kalayanarooj S, Charoensirisuthikul T, et al. Differences in global gene expression in peripheral blood mononuclear cells indicate a significant role of the innate responses in progression of dengue fever but not dengue hemorrhagic fever. The Journal of Infectious Diseases, (2008); 197(10): 1459-1467.
- Sun P, García J, Comach G, Vahey MT, Wang Z, et al. Sequential waves of gene expression in patients with clinically defined dengue illnesses reveal subtle disease phases and predict disease severity. PLoS Neglected Tropical Diseases, (2013); 7(7): e2298.
- Sim S, Hibberd ML. Genomic approaches for understanding dengue: insights from the virus, vector, and host. Genome biology, (2016); 17: 38.
- Nascimento EJ, Braga-Neto U, Calzavara-Silva CE, Gomes AL, Abath FG, et al. Gene expression profiling during early acute febrile stage of dengue infection can predict the disease outcome. PLoS One, (2009); 4(11): e7892.
- Castillo JA, Castrillón JC, Diosa-Toro M, Betancur JG, St Laurent G 3rd, et al. Complex interaction between dengue virus replication and expression of miRNA-133a. BMC Infectious Diseases, (2016); 16: 29.
- Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, et al. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Research, (2015); 43(Database issue): D153-D159.
- Josyula JVN, Talari P, Pillai AKB, Mutheneni SR. Analysis of gene expression profile for identification of novel gene signatures during dengue infection. Infectious Medicine, (2023); 2(1): 19-30.
- Zhong XL, Liao XM, Shen F, Yu HJ, Yan WS, et al. Genome-wide profiling of mRNA and lncRNA expression in dengue fever and dengue hemorrhagic fever. FEBS Open Bio, (2019); 9(3): 468-477.
- Bajrai LH, Sohrab SS, Alandijany TA, Mobashir M, Reyaz M, et al. Gene Expression Profiling of Early Acute Febrile Stage of Dengue Infection and Its Comparative Analysis With Streptococcus pneumoniae Infection. Frontiers in Cellular and Infection Microbiology, (2021); 11: 707905.
- Xie LM, Yin X, Bi J, Luo HM, Cao XJ, et al. Identification of potential biomarkers in dengue via integrated bioinformatic analysis. PLoS Neglected Tropical Diseases, (2021); 15(8): e0009633.
- Wickremsinghe IAC, Balasubramaniam V, Mot YY, Dhanoa A, S Hassan S. Identification of Differentially Expressed Genes in BALB/c Mouse Liver upon Primary Infection with DENV1 and Sequential Heterologous Infection with DENV2. Pathogens, (2018); 7(4): 78.
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Research, (2013); 41(Database issue): D991-D995.
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research, (2015); 43(7): e47.
- Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Research, (2023); 51(D1): D638-D646.
- Franz M, Lopes CT, Fong D, Kucera M, Cheung M, et al. Cytoscape.js 2023 update: a graph theory library for visualization and analysis. Bioinformatics, (2023); 39(1): btad031.
- Kolberg L, Raudvere U, Kuzmin I, Adler P, Vilo J, et al. g:Profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Research, (2023); 51(W1): W207-W212.
- Tokar T, Pastrello C, Rossos AEM, Abovsky M, Hauschild AC, et al. mirDIP 4.1-integrative database of human microRNA target predictions. Nucleic Acids Research, (2018); 46(D1): D360-D370.
- Han H, Cho JW, Lee S, Yun A, Kim H, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Research, (2018); 46(D1): D380-D386.
- Fan Y, Siklenka K, Arora SK, Ribeiro P, Kimmins S, et al. miRNet – dissecting miRNA-target interactions and functional associations through network-based visual analysis. Nucleic Acids Research, (2016); 44(W1): W135-W141.
- Cotto KC, Wagner AH, Feng YY, Kiwala S, Coffman AC, et al. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Research, (2018); 46(D1): D1068-D1073.
- Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Current Computer-Aided Drug Design, (2011); 7(2): 146-157.
- Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, et al. The Protein Data Bank. Acta Crystallographica. Section D, Biological Crystallography, (2002); 58(Pt 6 No 1): 899-907.
- Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. Journal of Chemical Information and Modeling, (2021); 61(8): 3891-3898.
- Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Systems Biology, (2014); 8 Suppl 4 (Suppl 4): S11.
- Grassi G. Metoprolol in the treatment of cardiovascular disease: a critical reappraisal. Current Medical Research and Opinion, (2018); 34(9): 1635-1643.
- Arakawa T, Kobayashi-Yurugi T, Alguel Y, Iwanari H, Hatae H, et al. Crystal structure of the anion exchanger domain of human erythrocyte band 3. Science, (2015); 350(6261): 680-684.
- Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. The Journal of Biological Chemistry, (2005); 280(10): 9595-9603.
- Liu Y, Yang J, Chen LM. Structure and Function of SLC4 Family HCO3- Transporters. Frontiers in Physiology, (2015); 6: 355.
- Yenchitsomanus PT, Kittanakom S, Rungroj N, Cordat E, Reithmeier RA. Molecular mechanisms of autosomal dominant and recessive distal renal tubular acidosis caused by SLC4A1 (AE1) mutations. Journal of Molecular and Genetic Medicine : An International Journal of Biomedical Research, (2005); 1(2): 49-62.
- Remigante A, Spinelli S, Pusch M, Sarikas A, Morabito R, et al. Role of SLC4 and SLC26 solute carriers during oxidative stress. Acta Physiologica (Oxford, England), (2022); 235(1): e13796.
- Kalli AC, Reithmeier RAF. Interaction of the human erythrocyte Band 3 anion exchanger 1 (AE1, SLC4A1) with lipids and glycophorin A: Molecular organization of the Wright (Wr) blood group antigen. PLoS Computational Biology, (2018); 14(7): e1006284.
- Stefanis L. α-Synuclein in Parkinson's disease. Cold Spring Harbor Perspectives in Medicine, (2012); 2(2): a009399.
- Bordbar S, Alijanzadeh D, Samieefar N, Khazeei Tabari MA, Pourbakhtyaran E, et al. The Role of Alpha-Synuclein in Neurodevelopmental Diseases. Molecular Neurobiology, (2024); 62: 962-972.
- Siddiqui IJ, Pervaiz N, Abbasi AA. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Scientific Reports, (2016); 6: 24475.
- van de Weg CA, van den Ham HJ, Bijl MA, Anfasa F, Zaaraoui-Boutahar F, et al. Time since onset of disease and individual clinical markers associate with transcriptional changes in uncomplicated dengue. PLoS Neglected Tropical Diseases, (2015); 9(3): e0003522.
- Hanley JP, Tu HA, Dragon JA, Dickson DM, Rio-Guerra RD, et al. Immunotranscriptomic profiling the acute and clearance phases of a human challenge dengue virus serotype 2 infection model. Nature communications, (2021); 12(1): 3054.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0![]()


