

Full Length Research Article
The development of cost effective 100 base pair prototype DNA ladder using polymerase chain reaction
Syed Moeezullah1, Anwar Khan2, Nusrat Jahan1*, Bushra Tabassum3, Inayat Agha1, Samia Parveen1, Muhammad Tariq Rao4, Zainia Rehmat5, Noreen Kasi6
Adv. life sci., vol. 10, no. 1, pp. 61-65, March 2023
*- Corresponding Author: Nusrat Jahan (Email: nusrat.jahanbuitems@gmail.com)
Authors' Affiliations
2. Department of Microbiology, Balochistan University of Information Technology, Engineering & Management Sciences (BUITEMS), Quetta – Pakistan
3. School of Biological Sciences, University of the Punjab, Lahore – Pakistan
4. National Centre of Excellence in Molecular Biology (CEMB), University of the Punjab, Lahore – Pakistan
5. Department of Biotechnology, SBKWU, Quetta – Pakistan
6. Department of Economics, Balochistan University of Information Technology, Engineering & Management Sciences (BUITEMS) Quetta– Pakistan
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: In genomics, DNA scale is used as a standard unit for the measurement of unknown DNA fragments, plasmids, and PCR products during gel electrophoresis. The 100 base pair DNA ladder is essential and cost-effective in molecular biological research and is available commercially which is too expensive and not easily accessible to a common researcher for laboratory usage.
Methods: The main purpose of this study was to report easily and practical method to prepare 100 base pair DNA ladder by simple PCR using pCAMBIA 1301 plasmid as a template which is an effective cost reduction strategy for laboratories. pCAMBIA 1301 was transformed into Escherichia coli (Top 10) bacteria by using heat shock method for high the yield of the plasmid. Bacteria containing our desire plasmid were cultured and plasmid was extracted from bacteria by using kit method. About 10 pairs of primers were designed from the backbone of the plasmid which amplifies 100 to 1000 base pair of PCR product with an interval of 100 base pair fragments. These fragments were optimized by using gradient thermo cycler and PCR products were purified using kit methods. For the stability of 100 base pair DNA ladder, it was placed in seven different buffers.
Results: The outcome of this study shown that polymerase chain reaction was able to amplify 10 different types of DNA fragments which ranges from 100 to 1000 base pair with high qualification and size accuracy. PCR products were purified and sequenced. DNA ladder was pooled in seven different buffers and stored at -20°C. These buffers were used to optimize and evaluate the stability of the prototype DNA ladder.
Conclusion: Our laboratory made 100base pair DNA ladder is very cost effective, it only cost 11 USD to prepare DNA ladder. This 100 base pair DNA ladder provides an independent quantitative unit that can be used with any biological application or technology, enabling genomes to be measured using a common metric.
Keywords: 100 bp DNA ladder, pCAMBIA 1301 plasmid; PCR technique; Gel electrophoresis; Break Even Point Analysis
Introduction![]()
In every field of science, scientific examinations depend on standard units for the measurement of quantitative descriptions of natural phenomena [1]. Scientists have established standard units for genomics and developed a DNA ladder that expresses the quantitative standard unit for the measurement of unknown DNA sequences. It acts as a scale that measures genetic features like human genetic variation and microbial variation [2]. The DNA ladder is a very important tool used in gel electrophoresis with DNA samples in the neighboring lanes to serve as references. An unknown DNA sample can be estimated by comparing it with the known bands of the DNA ladder on agarose gel or polyacrylamide gel [3]. According to the literature, numerous methods have been successfully applied to produce 100 base pair DNA ladders. Nowadays, DNA ladders are made in two ways: one by polymerase chain reaction amplification and the other by mixing PCR products at specific concentrations of fragments with definite sizes [4]. The second method involves the digestion of natural sources such as bacteriophages or plasmid DNA using restriction endonucleases [5]. For the construction of the DNA ladder, different bacterial plasmids, such as pCAMBIA, pTZ57R, and pMAL-c2X, are used as template DNA in PCR [6]. Both 100 base pair and 1 kb DNA ladders are produced using pairs of plasmids like pPSU1 and pPSU2. These plasmids can be isolated from 100 ml E. coli cultures, and they are also available without licensing, which provides researchers with high-quality, low-cost plasmids for molecular biological applications [1]. A 100 base pair DNA ladder has high economic importance as it is regularly used in research laboratories to carry out different experimental works [4]. Consequently, a DNA ladder has been prepared at a low cost. Comparing the commercial marker with the laboratory-made DNA ladder demonstrates the cost-effectiveness of laboratory production [7]. The 100 base pair DNA ladder is essential and cost-effective in molecular biological research, but it is often too expensive for laboratory usage and not easily accessible to the average researcher. Companies such as New England Biolabs (Canada) and Thermo Scientific (USA) offer 100 base pair DNA ladders at high costs, which can affect the financial resources of researchers. For specific requirements, a laboratory can produce a significant amount of DNA ladder, making it an effective cost-reduction strategy [8].
Methods![]()
Transformation and isolation
The transformation and isolation of the pCAMBIA 1301 plasmid was carried out as follows: In the PCR reaction, pCAMBIA 1301 plasmid was used as a template DNA. The plasmid was then transformed into Escherichia coli (Top 10) bacteria using the heat shock method as described by [9]. The transformed Escherichia coli (Top 10) bacteria containing our desired plasmid (pCAMBIA 1301) were cultured on LB agar (Luria-Bertani medium) plates containing Kanamycin antibiotic (50 µg/ml). A single colony of bacteria (transformants) was cultured again in LB broth containing Kanamycin antibiotic overnight. The next day, the pCAMBIA 1301 plasmid was purified using a kit method (Thermo Scientific GeneJET Plasmid Miniprep Kit #K0502, #K0503), and this plasmid was used as a template DNA in subsequent PCR reactions.
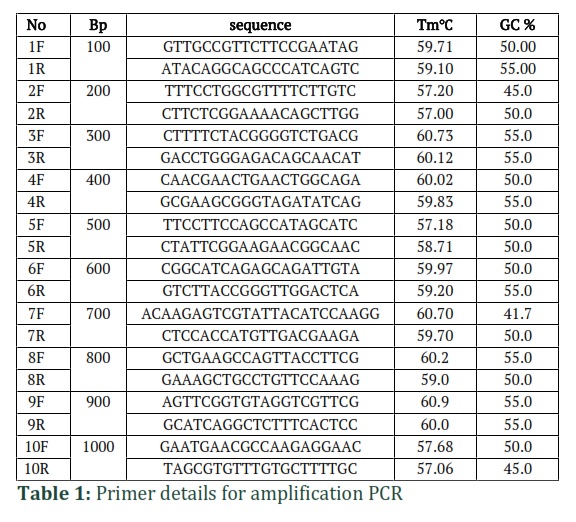
Designing of PCR primers. According to the plasmid DNA sequence (pCAMBIA 1301 plasmid), 10 pairs of primers were designed to amplify 100-1000 base pair fragments using the online tool Primer3Plus. These primers were purchased from Macrogen Company (Korea) for the successful amplification of the expected DNA fragments ranging from 100 to 1000 base pairs. The sequences and temperature of each primer are shown in Table 1, where "F" shows the forward strand, while "R" shows the reverse strand of the primer.
Optimization of fragments 100 base pair to 1000 base pair using gradient thermocycler
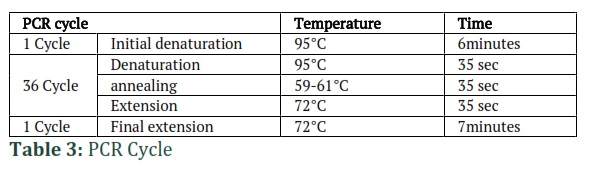
For each DNA fragment, a separate PCR reaction was performed in a 20 µl volume, containing 1.5 µl of the pCAMBIA plasmid template DNA, 2 µl of dNTP (2mM), 2 µl of Taq buffer (10x), 1.5 µl of MgCl2, 9.5 µl of PCR water (Thermo Fisher Scientific, USA), 3 µl of both forward and reverse primers (10 pmol each), and 0.5 µl of Taq DNA polymerase (5U/ µl, Thermo Fisher Scientific, USA). To amplify all DNA fragments at once, the optimal annealing temperature was determined using a temperature gradient ranging from 57-61°C. The PCR cycle program was designed in a thermal cycler with an initial denaturation step at 95°C for 6 min, followed by 36 cycles of denaturation at 95°C for 35 sec, annealing at 59-61°C for 35 sec (as illustrated in Table 1), and extension at 72°C for 35 sec. After the 36 cycles, a final extension was carried out at 72°C for 7 minutes.
PCR product purification
The PCR products of 100 to 1000 base pair were purified using the purification kit method (WizPrepTM Gel/PCR Purification Mini Kit) as instructed. Briefly, PCR products were treated with a PCR purification buffer (PCR purification buffer: PCR sample = 5:1). The mixture was transferred to the spin column and centrifuged at 14000 rpm for 1 minute. The flow-through after centrifuge was discarded and column re-inserted into the collection tube and samples were washed by 700 µl wash buffer (ethanol added) centrifuged for 30 sec. The supernatant was again discarded, and the spin column was centrifuged for 1 minute at 14000 rpm for the removal of residual Wash buffer completely. Spin column was placed in an autoclaved 1.5 ml tube and 40 µl elution buffer was added to the center of the spin column and placed for 1 to 2 minutes at room temperature and centrifuged at 14000 rpm for 1 minute for DNA elution.
Identification and sequencing of DNA fragments
A total of 4 µl (150 ng) of each PCR-purified product was sequenced on a 2% agarose gel stained with Ethidium Bromide (Thermo Fisher Scientific, USA). The products were then electrophoresed in 1X TBE buffer (Tris base, boric acid, EDTA) at 80 volts for 120 minutes, and the length of fragments was estimated by comparing to a 100 bp DNA size marker (Biolis Biodyne, 100 bp). The appearance of DNA bands was visualized under UV light at 260/280 nm on a gel documentation system. Purified DNA fragments were sequenced using the facilities of 1st Base Sequencing, Malaysia. The received sequences were analyzed using the Bio-Edit tool to compare the length and number of base pairs of fragments with the template DNA.
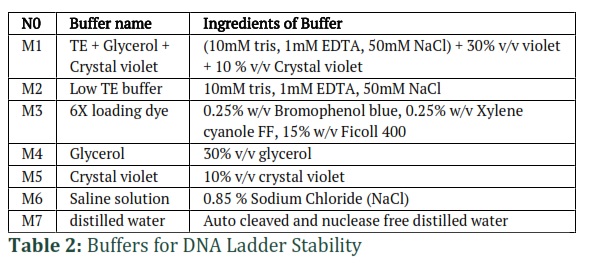
Stability and evaluation of 100base pair DNA ladder
Various sizes of purified PCR products were mixed according to the specific proportions and dissolved in seven different buffers (shown in Table 2) for DNA ladder stability. The ladder was ready for laboratory use and was compared with the commercial version. The 100 base pair DNA ladder was stored at -20°C. Finally, the cost-effectiveness of the 100 base pair DNA ladder was estimated using the Break-Even Point formula in Microsoft Excel.
Results![]()
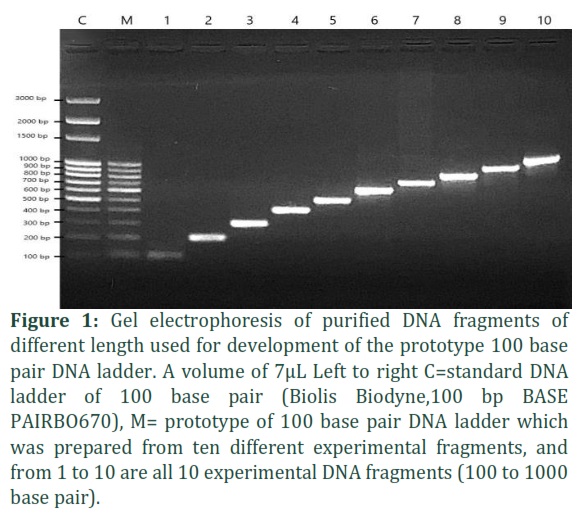
Amplification of PCR fragments using designed primers. The outcome of the use of the pCAMBIA 1301 plasmid as a template and its designed primers for the generation of 100 base pairs DNA ladder showed successful amplification of the expected (100 to 1000 base pair) DNA fragments (as shown in figure 1). The PCR reaction was performed in a single run PCR using the optimization process in temperature profile (See table 3). The DNA fragments were clearly observable on 1.7% agarose gel and size of all DNA bands, in comparison to the DNA marker or DNA ladder (Biolis Biodyne,100 BASE PAIRBO670) were closely related.
Preparation and stability of 100 base pair DNA ladder. Following extraction, purification and quantitative analysis, each fragment was quantified by using Nano drop. The PCR purified products as shown in figure 1 were mixed according to the special proportions, seven different buffers were used to evaluate the stability of the DNA fragments. The DNA fragments with buffer solution was then stored at -20°C for the further
testing procedure. The stability of DNA fragments was tested once a month post initial storage till about 6 months. The result of the agarose gel electrophoresis revealed that all the prototype100 base pairs DNA ladders bands were clear and our DNA ladder was comparable to a commercial and standard markers(See Figure 2).
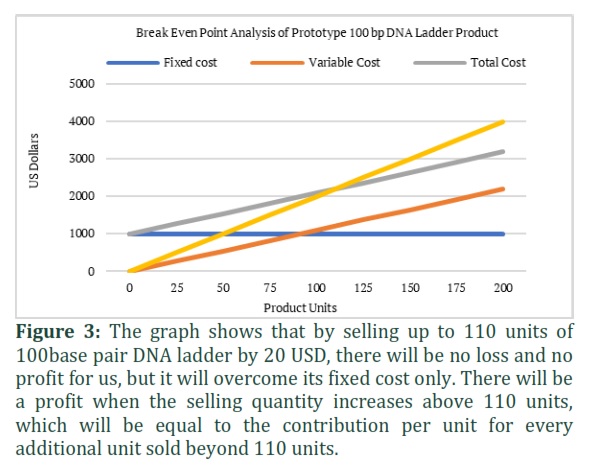
Cost effective strategy using Break Even Point analysis for production of 100 bp DNA ladder
A break-even point is a statistical analysis where the total costs (expenses) and total sales (revenue) of a product are equal. This means that there is neither profit nor loss at that point. In our study, we estimated the variable cost and fixed cost (capital cost) of DNA ladder production. We implemented the break-even point formula and found that if we sell a unit product for 20 US Dollars, there would be no profit and no loss to the laboratory after 110 units are sold.
Fixed cost = 995 USD
Variable cost per unit = 11 USD
Sale price per unit = 20 USD
Break Even Point (BEP)
BEP = (fixed cost)/(sales price-Variable cost)
BEP = 995/(20-11)
BEP = 110.55 units
Figures & Tables

The scientific examinations depend on standard units for the measurement of quantitative descriptions of natural phenomena. In molecular biology, scientists have established standard units for genomics to develop a DNA ladder that expresses the quantitative standard unit for the measurement of unknown DNA fragments by acting as a scale to measure genetic features like human genetic variation and microbial variation [10]. DNA ladder is essential and cost-effective in molecular biological research and is available commercially which is too expensive for laboratory usage. Therefore, the development of an indigenous DNA ladder can result in cost-effective strategies to curb expenses. In this study the main objective was to prepare cost-effective 100 base pair DNA ladder prototype with fragments of 100-1000 base pair with 100 base pair intervals.
For the development of the prototype plant expression vector, pCAMBIA 1301 was used as a templet. About 10 pairs of primers were designed from the backbone of the vector, ranging from 100 base pairs up to 1000 base pairs, with an interval of 100 base pairs. Each fragment was amplified using thermocycler and resolved on gel electrophoresis. The outcome of PCR product on agarose gel showed that all of the 10 experimental fragments (ranging from 100 to 1000 base pair) were exactly aligned with the respective bands of DNA ladder in the standard column as shown in the results. The fragments were subsequently purified and quantified using a nanodrop, then sequenced using the facility of 1st base Sequencing, Malaysia. Each purified fragment was pooled with seven different buffers and stored at -20°C.The stability of the ladder containing different buffers was evaluated on an agarose gel every 15 days up to 180 days. Further, costing was done using break even analysis.
Wang et al., prepared a DNA ladder with 12 fragments ranging from 100 to 1500 base pairs. The study resulted in the amplification of DNA fragments by PCR. Hekmatnezhad, et al., [14] also produced DNA ladders using different types of forward and reverse primers. The results of [11] showed successful amplification of DNA fragments ranging from 100 to 1000 base pairs in a single PCR reaction for the production of DNA ladder for various research purposes in molecular biology experiments. According to the researchers, the PCR product mixture was confirmed by agarose gel electrophoresis and used as a DNA ladder without any further purification. These unpurified PCR products were as reliable as markers from commercial sources. Similarly, [4] amplified 11 different DNA fragments ranging from 100 to 1,000 base pair, as well as a 1,500 base pair fragment, using PCR. Finally, [12] successfully produced and tested a standard ladder through PCR. Producing a 100 base pair DNA ladder through PCR is a simple, inexpensive, and time-saving method for laboratories.
Another technique for producing DNA fragments of various lengths is performed through Restriction enzymes (endonucleases). Henrici et al., conducted a study on pPSU1 & pPSU2 pairs of plasmids to produce 100 base pair and 1 kb DNA ladders by the digestion of two common restriction enzymes [15]. These pair of plasmids are used as reference fragments from 50 to 1000 base pair at a fraction of the cost of commercial DNA ladders. Hartley also invented the nucleic acid marker ladder through restriction endonuclease digestion, using restriction enzymes digestion a collection of DNA fragments resulting from complete digestion of one or more DNA fragments by one or more restriction endonucleases.
In our study, the purified DNA product of the DNA ladder was treated with various DNA stabilizers. Solutions with different concentrations, as cited in the literature, were used as stabilizing media for the DNA fragments. The solution containing DNA fragments was mixed with the stabilizing media and then stored at -20 °C for the further testing procedure. The stability of DNA fragments was tested for 6 months post initial storage.
Wang et al. [11] studied DNA fragment intensities, finding that the intensity of each fragment was proportional to its size and length, resulting in a lower intensity for the 100 base pair fragment compared to larger fragments. The researchers mixed different concentrations of DNA fragments to construct the DNA ladder for visualization. Similarly, Baoutina et al. stored 20 DNA fragments in TE buffer and distilled water at different temperatures, studying them for 10 weeks. Over this time period, 6 of the 20 DNA fragments dissolved in TE buffer stored at 25°C and 4 of the 20 fragments dissolved in distilled water stored at 4°C were degraded. However, the 20 DNA samples stored in TE buffer at 4°C remained stable for 10 weeks. Additionally, the DNA samples preserved at -20°C and -70°C were not degraded in either distilled water or TE buffer, even after 10 weeks. According to the study in [13], TE buffer is a good stabilizer for DNA samples, and can be used to preserve and store DNA fragments at -20°C or -70°C for an extended period.
In research, scientists use DNA ladder in the laboratory regularly for different purposes. However, commercial DNA ladders can be expensive and can affect the financial cost of researchers. Therefore, we developed a cost-effective prototype of a 100 base pair DNA ladder or marker using the polymerase chain reaction technique. We stored the prototype DNA ladder in different buffers to check its stability. Using our methodology, researchers can easily prepare a 100 base pair DNA ladder in the laboratory within a few days. This strategy is an effective way to reduce costs and improve the availability of DNA ladders for laboratories.
Author Contributions
All of the authors contributed in conception, acquisition of data analysis, design, drafting, interpretation of data and revising the article.![]()
The authors declare that there is no conflict of interest.
![]()
References
- Henrici RC, Pecen TJ, Johnston JL, Tan S. The pPSU plasmids for generating DNA molecular weight markers. Scientific reports, (2017); 7(1): 2045-2322.
- Reis AL, Deveson IW, Wong T, Madala BS, Barker C, et al. A universal and independent synthetic DNA ladder for the quantitative measurement of genomic features. Nature communications, (2020); 11(1): 1-11.
- Rashno M, Seyfi Abad Shapouri MR, Jolodar A. Construction of a synthetic vector for preparation of a 100 base pair DNA ladder. Iranian Journal of Biotechnology, (2012); 10(2): 106-110.
- Lertworapreecha M, Thongnan J. Construction of Recombinant DNA Plasmid for Production 100 Base Pair DNA Ladder for Endless Usage Based on PCR Technique. Genomics and Genetics, (2018); 11(1&2): 22-25.
- Chen Z, Wu J, Li X, Ye C, Wenxing H. Novel strategies to construct complex synthetic vectors to produce DNA molecular weight standards. Molecular biotechnology, (2009); 42(1): 128-133: 1073-6085.
- Saidijam M, Shahreza HK, Tehrani ZR, Karimizare S, Shabab N, et al. Designing and constructing an 100 bp DNA Ladder by combining PCR and enzyme digestion methods. Tehran University Medical Journal, (2011); 69(2).
- Kumar A, Singh S, Tomar A. Development of Midi-Plasmid Isolation Kit and Cost Optimization of DNA Molecular Weight Ladder: Economy of Laboratory vs. Commercial Products. Advances in Applied Science Research, (2010); 1(3): 129-146.
- Lan VTT, Loan PTT, Duong PAT, Thanh LT, Ha NT, et al. Straightforward procedure for laboratory production of DNA ladder. Journal of Nucleic Acids, (2012); 2012.
- Khan A, Nasir IA, Tabassum B, Aaliya K, Tariq M, et al. Expression studies of chitinase gene in transgenic potato against Alternaria solani. Plant Cell, Tissue and Organ Culture (PCTOC), (2017); 128563-576.
- Reis AL, Deveson IW, Wong T, Madala BS, Barker C, et al. A universal and independent synthetic DNA ladder for the quantitative measurement of genomic features. Nature Communications, (2020); 11(1): 3609.
- Wang TY, Guo L, Zhang J-h. Preparation of DNA ladder based on multiplex PCR technique. Journal of Nucleic Acids, (2010); 2010.
- Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR-driven overlap extension. Nature protocols, (2007); 2(4): 924-932.
- Baoutina A, Bhat S, Partis L, Emslie KR. Storage stability of solutions of DNA standards. Analytical chemistry, (2019); 91(19): 12268-12274.
- Hekmatnezhad H, Moradian F, Hashemi-Petroudi SH. Simple procedure for production of short DNA size markers of 100 to 2000 bp. Progress in Biological Sciences, (2017); 6(2): 199-204.
- Henrici RC, Pecen TJ, Johnston JL, Tan S. The pPSU plasmids for generating DNA molecular weight markers. Scientific reports, (2017); 7(1): 2438.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0


